
Abstract
The fall armyworm (Spodoptera frugiperda) is one of the most significant agricultural pests in the world and invaded China in early 2019. We sampled and sequenced RNA-seq data from 15 individuals across different developmental stages. Developmental stages were the larval stage (5th instar larvae and 6th instar larvae), chrysalis stage, and adult stage (female adult and male adult). Individual samples were mainly clustered by developmental stages and we then identified variation between developmental stages of differentially expressed transcripts (DETs). There were 2136 upregulated DETs and 1391 downregulated DETs in the larval stage when comparing larval and chrysalis stages. In the comparison between the chrysalis and adult stages, there were 2033 upregulated DETs and 1391 downregulated DETs in the chrysalis stage. In total, 19,195 abundantly expressed transcripts were obtained and 10% of them were DETs. We then obtained stage-specific DETs to investigate the potential function of the fall armyworm during different developmental stages. We also constructed our annotation background set for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. This indicated that the fall armyworm may undergo active metabolism during its lifespan, even in the chrysalis stage. And it also may experience detoxifying and xenobiotic metabolism throughout its life, especially in the larval stage, which partially explains the difficulty to eradicate using chemical control. Our study is the first insight into the developmental patterns of the fall armyworm and we also provide the fundamental information about enhanced drug resistance at the level of transcriptome. These results are beneficial for a future investigation related to the eradication and/or control stage.
Introduction
The fall armyworm (Spodoptera frugiperda) is one of the most significant agricultural pests in the world (Wang et al., 2018). It is native to Central and South America, and invaded China in early 2019 (Cui et al., 2019; Silver, 2019; Jing et al., 2020). Since then, the fall armyworm has expanded across China and its larvae have caused considerable damage to our agricultural production due to its rapid reproduction, dispersal, and migration capabilities. The fall armyworm life cycle length depends on the season, with 30 days in the summer and longer in the winter, and encompasses eggs, larval stage, chrysalis stage, and adult (Capinera, 1999). There are six instars in the larval stage, with natural molting, extensive feeding, and phenotypic changes (Capinera, 1999). In the chrysalis stage, it experiences active energy and nutrient metabolism because it does not enter diapause and continues morphogenesis and physiological activity (Johnson, 1987; Ramirez-Cabral et al., 2017). Reproduction normally takes about 1 week (Capinera, 1999).
Previous studies originally investigated the fall armyworm's life cycle, native distribution, host plants, and behavior as an agricultural pest (Foster, 1989; Capinera, 1999; Pannuti et al., 2016). Researchers then focused on its dispersal ability, environmental adaptability as well as its pesticide resistance, and detoxification (Itoh et al., 2018; Rodriguez et al., 2019; Wang et al., 2019; Liu et al., 2020; Nagoshi et al., 2020; Zhou et al., 2020). Researches then shifted to molecular studies, with the evaluation of larval gene expression differences on different host plants (corn and rice) to investigate the high plasticity in detoxifying-gene families of the fall armyworm (Silva-Brandao et al., 2017). After its Chinese invasion, the chromosome-scale genomes of the fall armyworm were reported describing gene variation, including the expansion of cytochrome P450 and glutathione s-transferase gene families (Liu et al., 2019a). A few months ago, mutations related to insecticide resistance were identified in the AChE gene in spreading Chinese populations (Zhang et al., 2020). Despite recent advancements in understanding the fall armyworm's ecology and genetics, our knowledge is limited regarding the developmental features of the fall armyworm and potential changes to insecticide resistance throughout its lifespan.
In this study, we focused on the developmental patterns of the fall armyworm using RNA-seq technology. There were 15 individuals sampled in our study, containing the larval stage (5th instar larvae and 6th instar larvae), chrysalis stage, and adult stage (female and male). We identified differently expressed transcripts (DETs) between different developmental stages and stage-specific DETs to investigate characteristic patterns of developmental changes, especially for detoxifying-related metabolism and changes in the basic metabolism.
Materials and Methods
Ethics statement
All the sampling processes in the study were approved by the Animal Ethics Committee of the College of Life Sciences of Sichuan University (Approval ID: 20210305001). All procedures were performed in accordance with the current laws of animal welfare and research in China.
Samples collecting
Fall armyworms were collected from the cornfield in Xindu District, Chengdu, Sichuan Province, China. They were bred in captivity and samples were collected from the first generation at the Institution of Plant Protection, Sichuan Academy of Agricultural Sciences. The temperature in the artificial box was 28°C ± 1°C. The relative humidity was 70% ± 10%. The light–dark schedule was 18:6, and the main ingredient of artificial feed was powdery corn. We sampled 5th instar larvae (N = 3), 6th instar larvae (N = 3), chrysalises (N = 3), female adults (N = 3), and male adults (N = 3). In total, there were 15 individuals sampled from three developmental stages. We removed intact larval intestinal tissues for sampling and sampled whole bodies from chrysalis and adult individuals. The sampled tissues were immediately mixed with liquid nitrogen, then stored at −80°C. We used TRIzol reagent (Invitrogen, Carlsbad, CA) to extract total RNA, and detected the RNA samples as follows: (1) analyze the degree of RNA degradation and contamination with agarose gel electrophoresis, (2) detect RNA purity (OD260/280 ratio) with Nanodrop, (3) qualify the RNA density with Qubit, and (4) analyze the RNA integrity with Agilent 2100.
Library construction and sequencing
After samples were qualified, mRNA was collected with Oligo dT enriching beads. Subsequently, a fragmentation buffer was added to break the mRNA into short fragments. Considering mRNA as a template, one-strand cDNA was synthesized with random hexamers, and buffer, dNTPs, and DNA polymerase I and RNase H were added to synthesize double-strand cDNA. Double-stranded cDNA was purified with AMPure XP beads. The purified double-stranded cDNA was subject to end-repair, poly (A) addition, and connected to the sequencing adapter, and then size selection was performed with AMPure XPbeads. Finally, libraries for samples were produced after performing PCR amplification, purifying the PCR products with AMPure XP beads.
After the libraries were constructed, we performed preliminary quantification with Qubit 2.0, diluting the library to 1.5 ng/μL, and then used Agilent 2100 to detect the insert size of the libraries. The library concentration was further detected with the Q-PCR method. Finally, it was sequenced with Illumina Hiseq 2000 in Novogene (Novogene, Beijing, People's Republic of China), and 150 bp paired-end (PE) raw reads data were generated for each library.
Transcript data processing
Raw reads were filtered to obtain clean reads as follows: (1) reads with adapters were removed, (2) reads with >10% N (N meant base were not identified) were removed, (3) low-quality reads were removed (reads with more than half bases with base quality Q-value ≤5), and (4) reads through NGS QC Toolkit v2.3.3 were filtered with strict thresholds of >90% reads with Q-value ≥20 (Patel and Jain, 2012). The subsequent analysis was performed with clean reads.
The fall armyworm reference genome and annotation files were downloaded from CNSA (CNGB Nucleotide Sequence Archive) database with project number CNP0000513. The clean reads were mapped to the reference genome with HISAT2 v2.1.0 (Kim et al., 2015), and the resulting SAM files were converted to BAM files, sorted by chromosome positions with Samtools v1.9 (Li et al., 2009). Finally, we used StringTie v1.3.64 to assemble transcriptomes and generate a GTF (general feature format) file containing expression information (Pertea et al., 2015). The
Functional annotation
The mRNA sequences of the fall armyworm were extracted from its reference genome based on the position annotation from the assembled GTF file. We used BLASTX to align the aforementioned mRNA sequence to Swiss-Prot and NCBI (
Differentially expressed transcript analysis
DETs between developmental stages were identified by DESeq2 in R packages (Love et al., 2014). The samples were grouped by developmental stages, containing larvae group (hereafter FL, consisting of 5th instar larvae and 6th instar larvae), chrysalis group (i.e., FC), and adult group (i.e., FA, three female, and three male adults). We compared FL versus FC, FC versus FA, and FL versus FA. We also obtained the stage-specific DETs by identifying the up- and downregulated DETs shared within each comparison. The p values were adjusted for multiple testing with the Benjamini–Hochberg (BH) method false discovery rate (FDR ≤0.05). Only transcripts that met FDR ≤0.05 and |log 2-fold change| ≥ 2 were identified as DETs.
Function enrichment analysis
The significant enrichment analysis of GO functions and KEGG pathways were used to identify the GO function terms and KEGG pathways that were significantly enriched in DETs. The biological functions of DETs that were significantly enriched were illustrated. We performed GO and KEGG enrichment analysis for DETs by utilizing the GO terms and KEGG pathways assigned by fall armyworm mRNA sequences as background data sets with clusterProfiler v3.16.0 R packages (Yu et al., 2012). In this study, the p values were also adjusted by the BH method, and only FDR ≤0.05 GO terms and KEGG pathways were significantly enriched.
Results
Transcriptome sequencing and alignment
We sampled 15 fall armyworm individuals from three developmental stages, 5th instar larvae, 6th instar larvae, chrysalis, female adults, and male adults to investigate the gene expression across developmental stages (Supplementary Table S1). The cDNA libraries for samples were sequenced by Illumina HiSeq 2000 platform and we obtained 406,595,447 raw reads and 122 GB data. After strict quality control, 398,109,548 (97.91%) clean reads were obtained and mapped to its reference genome. The mapping rates of 15 samples were between 77.98% and 86.31% (Supplementary Table S1). We performed the functional annotation of mRNA because of limited-feature annotation from the GTF file. In total, we obtained 13,511 GO terms and 7695 KEGG pathways for functional enrichment analysis.
Transcript expression analysis
Known transcripts in the fall armyworm reference genome with ≥5 reads in any sample were sufficiently abundant for further analysis. After filtering, 19,195 transcripts were abundantly expressed, accounting for 92.32% of known transcripts. The transcript expression matrix with reads count was normalized by DESeq2 (Love et al., 2014). Principal component analysis of the normalized expression matrix showed that fall armyworm larvae, chrysalis, and adult groups were separate. Samples were segregated by group on PC1, which explained 23.3% of the variance (Fig. 1). There were also significant differences in gene expression between larval stages and between genders.

The principal components analysis of three developmental stages in the fall armyworms. The expression of transcripts was transformed using rlog function.
The developmental difference in fall armyworms
DETs were identified according to chronological developmental stages. When comparing FL versus FC, there were 2136 upregulated DETs and 1391 downregulated DETs in the FL group. There were 2033 upregulated DETs and 1391 downregulated DETs in the FC group when comparing FC versus FA (Fig. 2).
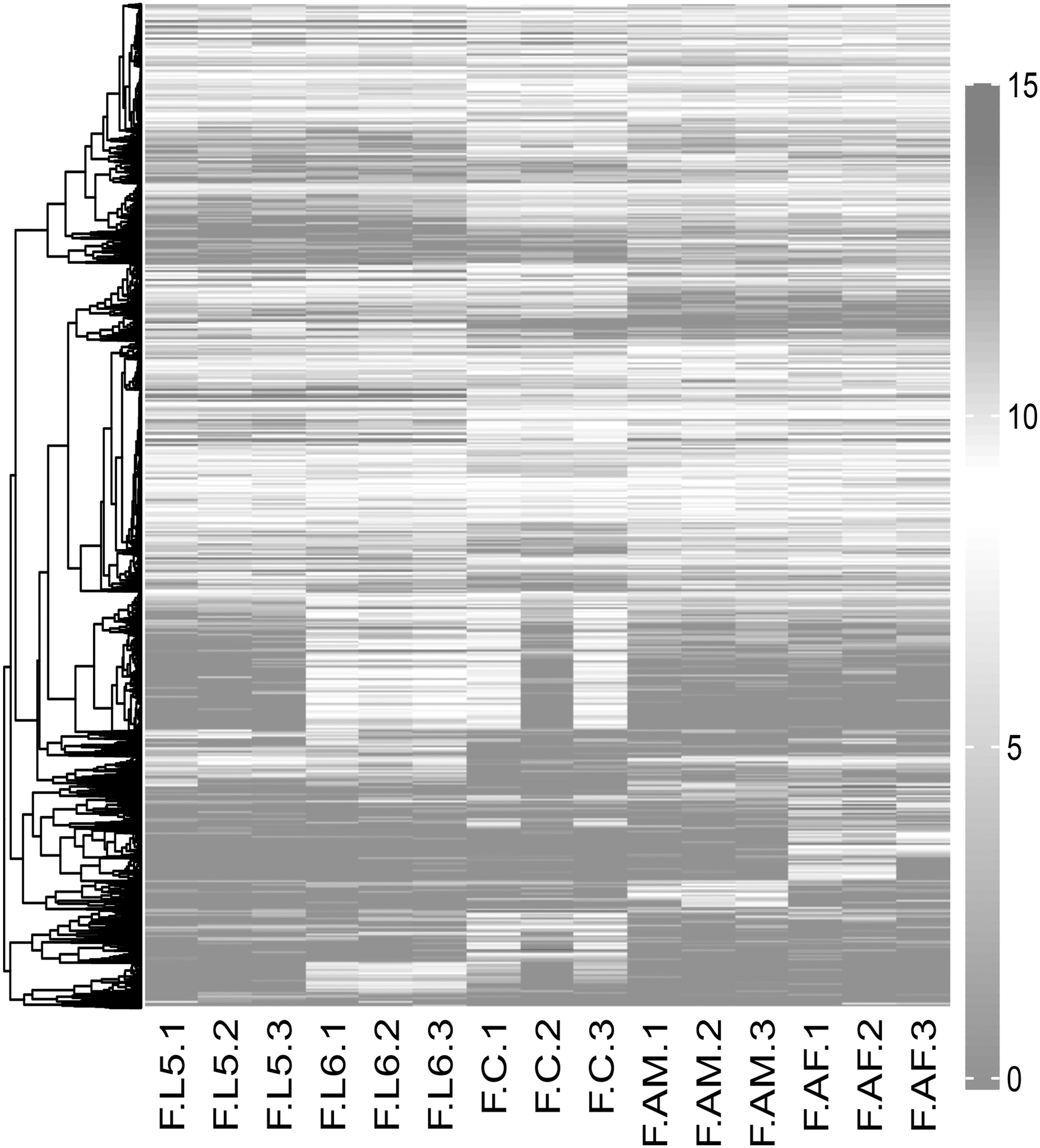
The heat map of expression of all DETs identified by differentially expressed analysis. The expression of DETs was normalized using rlog function.
We then performed the GO and KEGG enrichment analysis to investigate the biological functions of DETs during the different developmental stages. Upregulated DETs in the FL group (i.e., FL vs. FC) were mainly enriched in biological process (BP) and molecular function (MF) GO terms, including biosynthesis-related process and enzyme-related function (Supplementary Table S2). For KEGG analysis, many metabolism-related KEGG pathways were also enriched by these DETs, such as porphyrin and chlorophyll metabolism (ko00860) and drug metabolism—cytochrome P450 (ko00982) (Supplementary Table S2). Downregulated DETs in the FL group were mainly enriched in nervous-system-related BP terms, such as neuron cell–cell adhesion (GO:0007158) and presynaptic membrane assembly (GO:0097104). Many binding-related MF terms were enriched by these DETs, such as heparin binding (GO:0008201), roundabout binding (GO:0048495), and chitin binding (GO:0008061) (Supplementary Table S2). There were only two KEGG pathways, axon guidance (ko04360) and cell adhesion molecules (CAMs) (ko04514), which were enriched by these downregulated DETs (Supplementary Table S2).
Upregulated DETs in the FC group (i.e., FC vs. FA) were mainly enriched in metabolic and developmental BP terms, such as chitin catabolic process (GO:0006032), spermatid development (GO:0007286), and sensory perception of sound (GO:0007605) (Supplementary Table S3). Basic biosynthesis and metabolism-related KEGG pathways were also enriched such as arginine biosynthesis (ko00220), carbon metabolism (ko01200), and citrate cycle (TCA cycle) (ko00020) (Supplementary Table S3). Downregulated DETs in the FC group were mainly enriched in MF and BP terms that were associated with enzyme activity and behavior, such as acid phosphatase activity (GO:0003993), oxidoreductase activity (GO:0016491), chorion-containing eggshell formation (GO:0007304), and positive phototaxis (GO:0046956) (Supplementary Table S3).
In summary, we detected many BPs, including basic metabolism, detoxifying and xenobiotic metabolism, and developmental processes in these comparisons. We also found four GO terms and 14 KEGG pathways enriched by upregulated DETs in both FL versus FC and FC versus FA comparisons (Figs. 3, 4, and Supplementary Table S4). There was one GO term (GO:0004252) enriched by downregulated DETs in both FL versus FC and FC versus FA comparisons.
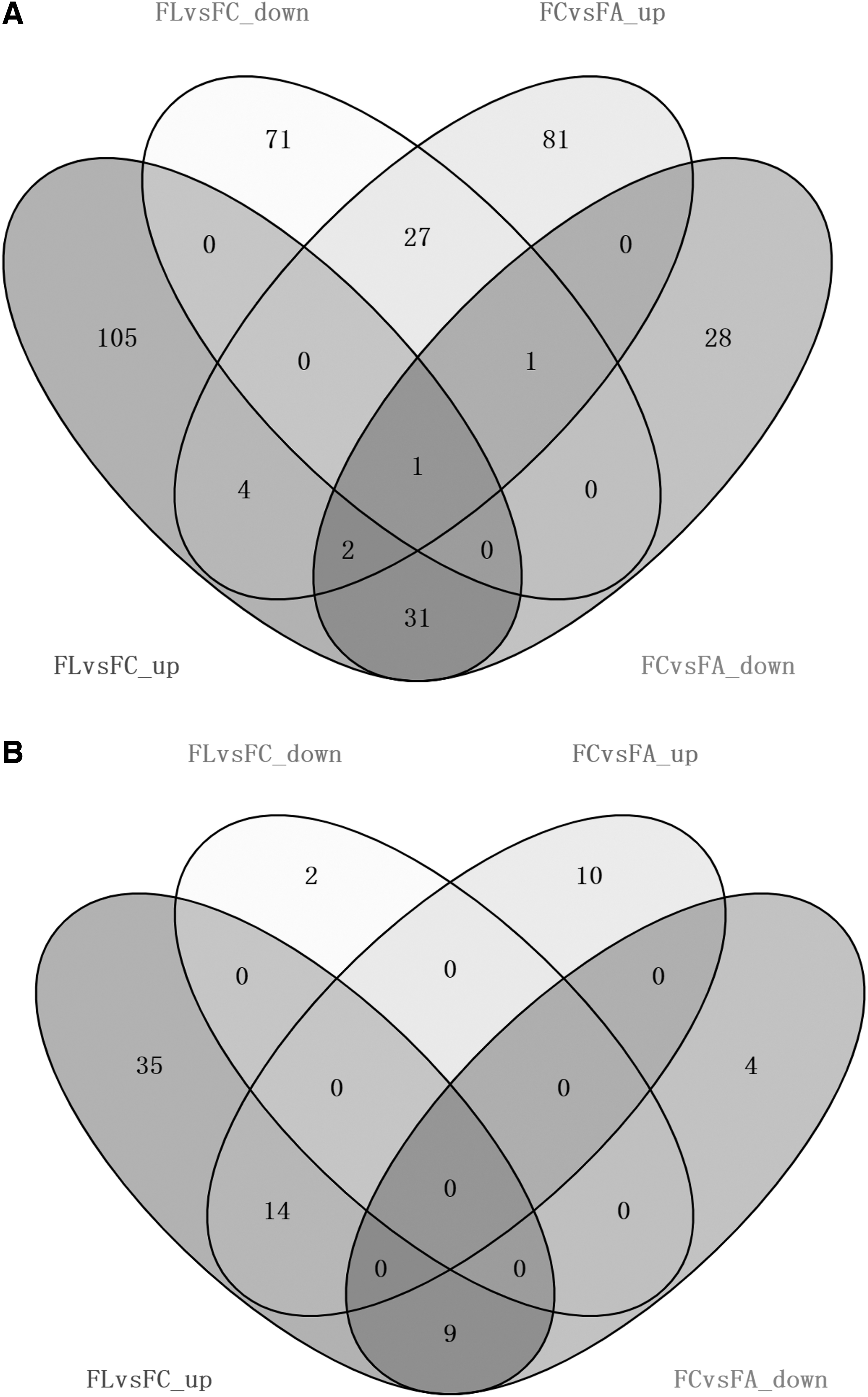
The Venn figure of GO and KEGG annotation of DETs in FL versus FC and FC versus FA. The GO terms
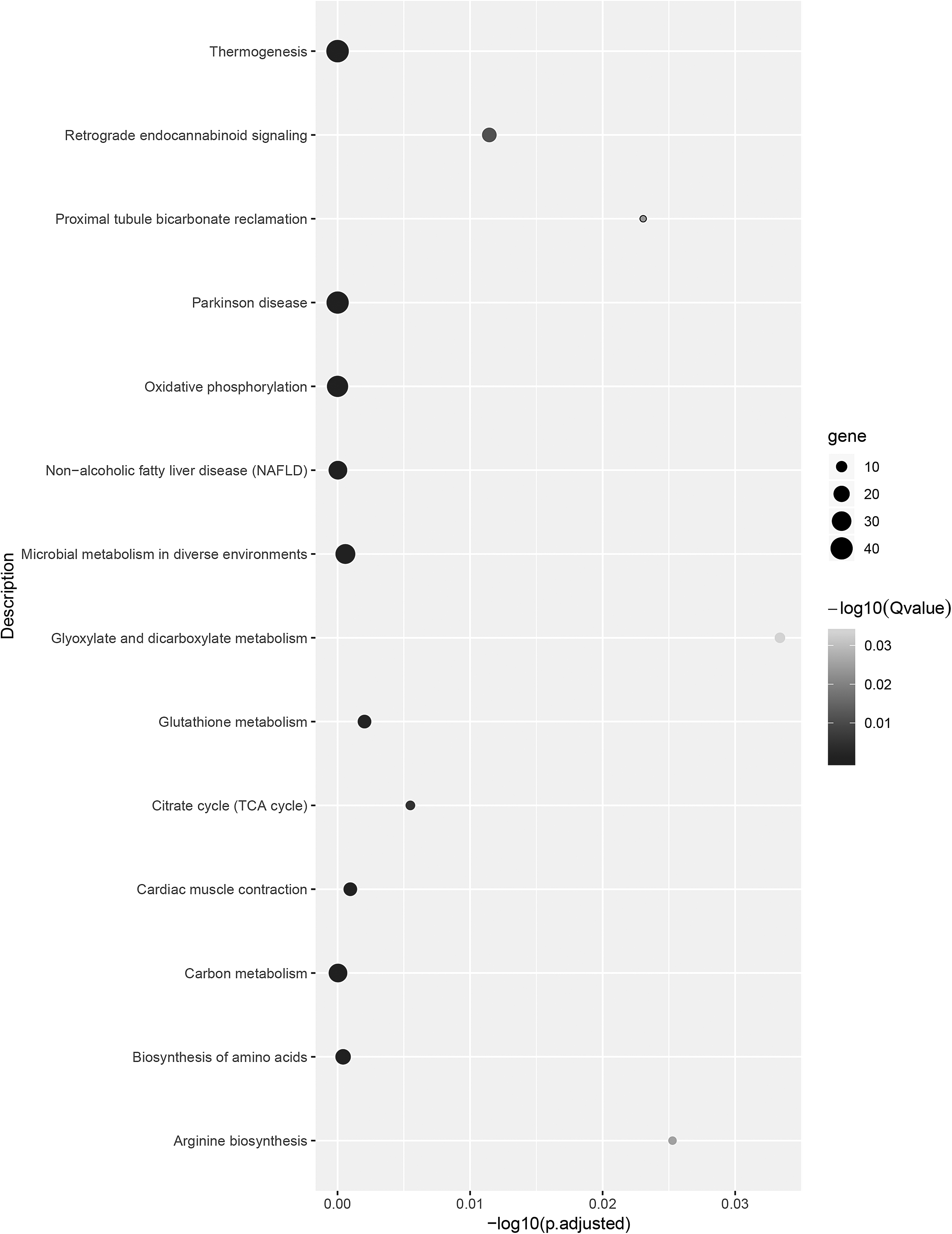
The 14 KEGG pathways enriched by upregulated DETs in the larvae stage (FL vs. FC) and in the chrysalis stage (FC vs. FA).
The stage-specific differences in fall armyworms
Stage-specific up- and downregulated DETs were identified in the three developmental stages. There were 1024 upregulated and 557 downregulated stage-specific DETs in the larval stage. The GO and KEGG enrichment analysis showed that upregulated larval stage-specific DETs were mainly enriched in energy and nutrient metabolism-related BP terms, as well as enzyme and binding-related MF terms, including chitin metabolic process (GO:0006030), iron ion binding (GO:0005506), and scavenger receptor activity (GO:0005044) (Supplementary Table S5). Drug metabolism-related KEGG pathways were enriched in the larval stage, such as drug metabolism—cytochrome P450 (ko00982) and metabolism of xenobiotics by cytochrome P450 (ko00980). Furthermore, there were few GO terms and KEGG pathways that were enriched by downregulated larval stage-specific DETs. For example, there was only one enriched KEGG pathway, lysosome (ko04142) (Supplementary Table S5).
In the chrysalis stage, there were 613 upregulated and 899 downregulated stage-specific DETs. The GO and KEGG enrichment analysis showed that upregulated chrysalis stage-specific DETs were mainly enriched in developmental related BP terms, such as cuticle pattern formation (GO:0035017), animal organ morphogenesis (GO:0009887), and nervous system development (GO:0007399) (Supplementary Table S6). There were only two KEGG pathways enriched by these DETs, axon guidance (ko04360), and Hippo signaling pathway—fly (ko04391). The MF GO terms were mainly enriched by downregulated chrysalis stage-specific DETs, such as iron ion binding (GO:0005506) and mono-oxygenase activity (GO:0004497) (Supplementary Table S6). The biosynthesis-related KEGG pathways associated with these DETs included drug metabolism—other enzymes (ko00983) and carbohydrate digestion and absorption (ko04973).
There were 1035 upregulated and 1298 downregulated adult stage-specific DETs. The GO and KEGG analysis showed that these upregulated DETs were mainly enriched in metabolic GO terms such as digestion (GO:0007586), and three KEGG pathways, nonhomologous end-joining (ko03450), neuroactive ligand-receptor interaction (ko04080), and lysosome (ko04142) (Supplementary Table S7). The GO annotation analysis of downregulated adult stage-specific DETs identified terms that were mainly enriched in, for example, multicellular organism development (GO:0007275), sperm axoneme assembly (GO:0007288), and axoneme assembly (GO:0035082). The metabolic KEGG pathways were associated with these downregulated DETs included the citrate cycle (TCA cycle) (ko00020) and glutathione metabolism (ko00480) (Supplementary Table S7).
Discussion
The fall armyworm is a significant agricultural pest across the world but has only recently invaded China with considerable expansion in its first 2 years (Wang et al., 2018; Jing et al., 2020). It has been suggested that eradication in China is impossible because of its enhanced drug resistance and reproductive ability (Silver, 2019; Zhang et al., 2020). The fall armyworm is experiencing gene expansion promoted by manual intervention and speciation affected by feeding (Liu et al., 2019a; Zhang et al., 2020; Zhou et al., 2020). Most studies have focused on adaptive evolution identifying larval gene expression (Silva-Brandao et al., 2017; Liu et al., 2019a), whereas a few have focused on its developmental patterns (Gu et al., 2013; Hardke et al., 2015). Our study sampled and sequenced RNA data from 15 fall armyworm individuals across three developmental stages. We obtained 19,195 abundantly expressed transcripts and about 10% were DETs. We also constructed our annotation background set for GO and KEGG enrichment analysis. Many basic metabolisms and xenobiotic BPs were identified in the fall armyworm's developmental stages.
The fall armyworms may possess the ability to activate xenobiotic and detoxifying metabolism throughout their lifespan and it is most active in the larval stage. First, we identified DETs between developmental stages and stage-specific DETs, and these transcripts were involved in detoxifying and xenobiotic metabolism. KEGG pathways enriched by upregulated DETs in the larval stage (FL vs. FC) were associated with many detoxifying and xenobiotic pathways, including glycine, serine, and threonine metabolism (ko00260), glutathione metabolism (ko00480), porphyrin and chlorophyll metabolism (ko00860), drug metabolism—other enzymes (ko00983), and drug metabolism—cytochrome P450 (ko00982). Furthermore, one of the shared upregulated KEGG pathways in the larval (FL vs. FC) and chrysalis stages (FC vs. FA) was glutathione metabolism (ko00480) (Fig. 4).
Second, MGST1 (microsomal glutathione S-transferase 1, XP_026744392.1-D2) and GstS1 (glutathione S transferase S1, XP_026313611.1) that provide glutathione peroxidase activity (Bresell et al., 2005) were upregulated in the larval (FL vs. FC) and chrysalis stages (FC vs. FA). Glutathione plays an important role in maintaining a physiological balance between prooxidants and antioxidants, crucial for pesticide resistance (Casida, 2017; Silva-Brandao et al., 2017). Besides, serine previously shown upregulation is a substrate for glutathione synthesis (Rodriguez et al., 2019).
Third, unlike in the adult stage, glutathione metabolism was upregulated in the larval and chrysalis stages, and the greatest number of detoxifying KEGG pathways was in the larval stage. Therefore, we speculate that fall armyworms possess the ability to activate xenobiotic and detoxifying metabolism throughout its lifespan, but it is most active in the larval stage. The fall armyworm is a polyphagous insect and is exposed to pesticides throughout its lifespan, which necessitates its high resistance (Wang et al., 2018; Zhang et al., 2020). Hence, the ability to develop the P450 system and to break down porphyrin is a growing important detoxification system in herbivores (Martinez et al., 2017; Itoh et al., 2018), and in our study, upregulated genes within this ability were identified throughout its lifespan. This may entirely or partially explain the difficulty to eradicate using chemical control. The fall armyworm invaded China 2 years ago, which places significant pressure on successful eradication and/or control (Cui et al., 2019; Wu, 2020).
Numerous basic metabolism was detected during developmental stages, and in the chrysalis stage, there were upregulated BPs related to reproductive organs and organ development. Through GO and KEGG enrichment analysis, we found that the biological functions were mainly related to energy metabolism in all developmental stage comparisons, and the developmental processes were upregulated in the chrysalis stage. For example, there were four pathways upregulated in the chrysalis stage (FC vs. FA) being the citrate cycle (TCA cycle) (ko00020), oxidative phosphorylation (ko00190), glyoxylate and dicarboxylate metabolism (ko00630), and thermogenesis (ko04714) (Fig. 4). Furthermore, the cbd-1 (Chitin-binding domain protein cbd-1, XP_026741077.1-D3) was upregulated in the larval and chrysalis stage. Chitin is key in the formation of the exoskeleton and the peritrophic matrix, which is closely related to the growth and development. It facilitates complex structures involving different cuticle types and could be modified at every developmental stage (Zhu et al., 2016; Liu et al., 2019b). The fall armyworm lifespan is a nondiapause insect with about a month life circle under optimum breeding conditions (Johnson, 1987; Ramirez-Cabral et al., 2017; Stokstad, 2017). Hence, it seems reasonable that we detected numerous basic metabolisms from the larval to the adult stage.
Conclusion
In summary, we sampled and sequenced RNA data from 15 fall armyworm individuals over the three developmental stages. We obtained 19,195 abundantly expressed transcripts and 10% were DETs. Unlike other complete metamorphosis insects within dormancy, basic metabolism and organ morphogenesis were rather active in the chrysalis stage. Moreover, the xenobiotic and detoxifying ability were most abundant in the larval stage, even though the fall armyworm had the potential for this ability throughout its lifespan. Our study is the first insight into the developmental patterns of the fall armyworm and we also provide the fundamental information about enhanced drug resistance at the level of transcriptome. These results are beneficial for a future investigation related to the eradication and/or control stage.
Footnotes
Authors' Contribution
B.Y. designed the study and collected data. R.T., X.L., and L.W. analyzed the data. Q.Y. and L.W. integrated and wrote the article. Z.F., J.L., X.L., M.P., and B.Y. provided resources and edits to article drafts. All authors revised the article and agreed with its publication.
Disclosure Statement
The authors declare that they have no competing interests, including specific financial interests and relationships and affiliations relevant to the subject of the article, and all authors have seen and agreed with the contents of the article.
Data Availability Statement
The data have been deposited in the CNGBdb database, the project number is CNP0001314.
Funding Information
This study was supported by Sichuan Key R&D Program (2019YFN0180).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.