
Abstract
Hydrogen sulfide (H2S) has been reported to participate in intestinal mucosal defense and repair. However, the precise regulatory mechanisms of H2S in ulcerative colitis (UC) remain unclear. We explored the effects of sodium hydrosulfide (NaHS), a donor of H2S, in dextran sulfate sodium (DSS)-induced colitis in rats. The pathologic features were determined by analyzing the hematoxylin and eosin-stained samples. Interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and myeloperoxidase (MPO) levels were determined using ELISA. The presence of cystathionine-γ-lyase (CSE) and light chain 3B (LC3B) were determined using immunohistochemical and immunofluorescence (IF) approaches, respectively. Next, we investigated the effects of NaHS in lipopolysaccharide (LPS)-stimulated human colonic smooth muscle cells (H2940). The level of reactive oxygen species (ROS) was determined using IF. NOD-like receptor 3 (NLRP3) and CSE were detected using western blot and quantitative real-time polymerase chain reaction. Autophagy was determined using western blot, IF, and electron microscopy. NaHS treatment considerably diminished colitis-induced histological injury and proinflammatory cytokine expressions. MPO, CSE, and H2S were downregulated, whereas LC3B was upregulated after NaHS administration in colitic rats. NaHS remarkably attenuated the levels of ROS, CSE, and NLRP3 in LPS-stimulated cells and enhanced autophagy, as was revealed by increased LC3-II-to-LC3-I ratio, elevated LC3, and decreased p62. Importantly, NaHS promoted autophagosome formation in LPS-treated cells. Exogenous H2S ameliorates intestinal injury by downregulating inflammation and activation of autophagy, suggesting the potential of NaHS as a therapeutic agent for UC.
Introduction
Crohn's disease and ulcerative colitis (UC) are inflammatory bowel diseases (IBDs) characterized by chronic, idiopathic gastrointestinal inflammation (Rosen et al., 2015; Ramos and Papadakis, 2019). UC has a multifactorial pathogenesis. Genetic susceptibility, defects in epithelial barrier function, dysregulation of the immune system, and environmental factors (Ungaro et al., 2017) are the primary risk factors, although the exact disease mechanism remains unclear. Recently, therapeutic approaches mainly focusing on immunosuppression and anti-inflammation have effectively improved patient outcomes (Panes and Alfaro, 2017). However, failure to achieve full disease remission and high recurrence risk is not uncommon (Beaugerie et al., 2020). It is, therefore, important to explore novel therapeutic strategies to address the limitations of the currently available treatment approaches.
Hydrogen sulfide (H2S), a gaseous messenger molecule in mammals, is predominantly synthesized from L-cysteine catalyzed by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (Paul and Snyder, 2012; Kimura, 2014). H2S can affect different bodily systems, such as the cardiovascular, neuronal, and gastrointestinal systems, in addition to its pleiotropic effects on physiology (Singh and Lin, 2015).
Previous research has demonstrated that sodium hydrosulfide (NaHS), an H2S donor, suppressed aspirin-induced leukocyte adherence in mesenteric venules and inhibited leukocyte infiltration in an air pouch model, indicating its modulating effects in leukocyte-mediated inflammation (Zanardo et al., 2006). Intravenous administration of NaHS reduced histological injury and alleviated proinflammatory cytokines in the dextran sulfate sodium (DSS)-induced colitis mouse model (Chen and Liu, 2016). However, the exact effect and action mechanism of NaHS in UC are unclear.
Inflammation and inflammatory cytokines appear to play essential roles in the initiation and development of UC (Yao et al., 2019). Cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6, serve as upstream facilitators and downstream mediators of inflammation (Strober and Fuss, 2011; Fonseca-Camarillo and Yamamoto-Furusho, 2015). Inflammasomes are part of innate immunity involved in inflammatory responses.
The inflammasome intracellular NOD-like receptor 3 (NLRP3) plays a crucial role in regulating intestinal homeostasis. In the presence of microbial ligands, the innate immune receptor mediates the inflammasome complex assembly, promoting caspase-1 activation and IL-1β secretion (Zaki et al., 2011). Interestingly, the downstream effector proteins have been shown to have both detrimental and protective roles in mucosal immunity (Zhen and Zhang, 2019). NaHS attenuated DSS-induced colon injury through NF-κB activation suppression both in vivo and in vitro (Chen and Liu, 2016), implicating its positive role. However, whether NaHS affects the NLRP3 inflammasome, involved in colon injury, and UC-induced inflammatory cytokines remains undetermined.
Autophagy is a cellular stress-response catabolic mechanism. It protects cells by removing the damaged organelles and proteins from the cytosol (Dikic and Elazar, 2018). The well-regulated, protein conjugation-dependent process plays a significant role in cell homeostasis (Noh et al., 2016). In the process, the autophagic adaptor protein p62 binds to the microtubule-associated protein 1 LC3 (Saikh et al., 2019), facilitating the transport of target substrates to the autophagosome for degradation. Binding of LC3 to phosphatidylethanolamine leads to the formation of LC3-II, which is presented to autophagosomes for elimination (Cha-Molstad et al., 2017). Therefore, the amount of LC3-II reflects the magnitude of autophagy induction.
Impaired epithelial autophagy is potentially implicated in IBD pathogenesis (Wu et al., 2018), indicating the value of modulating the immune process to treat the disease (Angelidou et al., 2018; Takagawa et al., 2018). It is also worth exploring whether NaHS modulates autophagy during colitis initiation and development.
In this study, we explore the effects of NaHS in a DSS-induced colitis rat model, which mimics human UC, and in lipopolysaccharide (LPS)-stimulated human colonic smooth muscle cells (H2940) in vitro. We verified whether NaHS affects colon injury, expression of inflammatory and autophagy factors, and autophagosome formation. We propose a new therapeutic strategy for UC based on the anti-inflammatory and pro-autophagy effects of NaHS.
Materials and Methods
Animals
In total, 18 adult male Sprague Dawley rats, each weighing 180–200 g, were obtained from the Guilin Medical College Experiment Center. The animals were acclimatized under controlled conditions of humidity (45% ± 10%), room temperature (23°C ± 1°C), and light/dark cycle (12 h/12 h) for 7 days before the start of the experiments. The rats had free access to water and commercial feed during the experimental period. The experimental protocol was approved by the Institutional Animal Ethics Committee and Animal Care Guidelines for the Care and Use of Laboratory Animals of Guilin Medical University (G20190302).
Reagents
NaHS and LPS were obtained from Acros Organics (Loughborough, UK). DSS was acquired from AMRESCO, Inc. (Solon, OH). The following antibodies were used in the experiment: anti-CSE (1:500; Sigma), anti-NLRP3 (1:500; Sigma), anti-LC3B (1:500; Abcam), anti-p62 (1:500; Thermo), anti-beclin 1 (1:500; Thermo), and anti-GAPDH (1:1000; Millipore).
Rat model of UC
The rats were randomly allocated to three groups, with six animals in each group. The control group was comprised of healthy rats administered with commercial feed. The UC group animals were administered DSS (2% w/v) and drinking water for 7 days. The NaHS group rats were treated with daily intraperitoneal injections of the compound at 2 mg/kg/day for 7 days after UC induction. NaHS hydrolyzes to spontaneously form H2S in aqueous solution; the equations are as follows: NaHS→Na+ + HS–; 2HS–→H2S + S2–; HS– + H+→H2S.
Cell culture
Human colonic smooth muscle cells (H2940) were obtained from Procell (Shanghai, China) and cultured in the DMEM supplemented with 10% fetal bovine serum (Gibco), 1% penicillin (Abcam), 1% streptomycin (Abcam), and 1% NEAA (Abcam) under 5% CO2 at 37°C. The model group cells were treated with LPS (10 μg/mL; Sigma Aldrich, St. Louis, MO). The NaHS group was treated with LPS (10 μg/mL), followed by NaHS.
Tissue sample collection
On day 7, animals were anesthetized using sodium pentobarbital (100 mg/kg). The colon tissues were collected immediately and washed with phosphate-buffered saline (PBS) and stored at −80°C or treated with 10% formalin in PBS solution for 24 h. They were then used for pathological analysis.
H2S determination
The proximal colon tissues were placed in a saline solution containing L-cysteine and pyridoxal phosphate after removing mucosa and submucosa. The produced H2S was added into the solution of zinc acetate (1%) to generate zinc sulfide. The mixture was allowed to stand at room temperature for 5 min, followed by the addition of trichloroacetic acid to stop the reaction. Next, the mixture was added to the hydrochloric acid solution containing N-n-dimethyl-p-phenylenediamine sulfate and FeCl3. The absorbance of the mixture was read at 670 nm, and H2S concentration was estimated using the standard H2S curve.
ELISA
According to the manufacturer's instructions, ELISA quantitative kits (Elabscience Biotechnology Co., Ltd., Wuhan, China) were used to assess the plasma concentrations of IL-1β, IL-6, and TNF-α. The mucosal layer and mucosal myeloperoxidase (MPO) activity of the colonic tissue samples were determined using the ELISA kits.
Hematoxylin and eosin staining and determination of histological score
The hematoxylin and eosin (HE)-stained, paraffin-embedded sections were submitted for histological analysis. Briefly, tissues (1 cm) harvested from the distal third of the colon were fixed in formalin (4%) for 24 h. Next, paraffin was applied to embed the colonic tissues, followed by HE staining. The histological score was assessed with the infiltration score and epithelial damage, and each slide was evaluated independently by two blinded researchers.
Immunohistochemistry
Tissues (1 cm) harvested from the distal third of the colon were fixed in formalin (4%) for 24 h and embedded using paraffin. Additionally, the tissues were treated with 3% hydrogen peroxide to suppress the endogenous peroxidase activity. The tissues were first treated with monoclonal anti-CSE for 4°C overnight, followed by the addition of the corresponding secondary antibody at 37°C for 30 min. The sections were then treated with 3,3′-diaminobenzidine-tetrahydrochloride before analysis.
Immunofluorescence
The tissue sections were successively deparaffinized and heat-fixed. The sections were washed with PBS three times after incubating with anti-LC3B, anti-reactive oxygen species (ROS), and anti-LC3 antibodies (1:100) overnight. Next, the sections were incubated with the corresponding secondary antibody (BOSTER) for 40 min. The sections were treated with fluorescein (1:100) for 1 h and washed again with PBS. Finally, all the sections were counterstained with Prolong™ Diamond Antifade Mountant with DAPI and sealed with glass coverslips. Immunofluorescence (IF) images were obtained using a fluorescence microscope.
Quantitative real-time polymerase chain reaction
The obtained colonic tissues were frozen immediately using liquid nitrogen and stored at −80°C until use. Complementary DNA was synthesized using 1.5 mg mRNA and reverse transcribing using PrimeScript™ RT Master Mix (TaKaRa Biotechnology), according to the standard protocols. qPCR SYBR Green Master Mix (Agilent Technologies, Santa Clara, CA) was used for quantitative real-time polymerase chain reaction (qRT-PCR). The relative expressions were standardized to GAPDH, and each measurement was obtained three times. The sequences of the primers used in this experiment are listed in Table 1.
Sequences of Primers Used in Quantitative Real-Time Polymerase Chain Reaction
Western blot
Colonic proteins were extracted using the RIPA buffer, and protein concentrations were measured using the BCA protein assay kit (Thermo). Next, 50 μg of the total protein from each sample was loaded and separated using 15% SDS-PAGE. The gels were then transferred to the PVDF membranes (Millipore). To block the nonspecific binding sites, 5% skim milk was applied to the membranes and incubated at room temperature for 1 h. The membranes were then incubated with the primary antibodies (anti-NLRP3, anti-CSE, anti-LC3-II, anti-LC3-I, anti-p62 anti-beclin 1, and anti-GAPDH) at 4°C overnight and secondary antibodies at room temperature for 1 h, successively. The ECL detection reagents (Abcam, Cambridge, MA) were used to visualize the blots. The density value of each band was measured using ImageJ software, and data were normalized with GAPDH. The relative expressions of each protein were presented as the ratio of the density value between the experimental and control samples. Each experiment was repeated three times.
Electron microscopy
Samples measuring >1 mm3 were separated and fixed using glutaraldehyde at 4°C for 4 h. Next, the samples were dehydrated using a graded series of ethanol and propylene oxide. They were then embedded in epoxy resin and submitted to transmission electron microscopy (Hitachi, Tokyo, Japan).
Statistical analysis
The results are presented as mean ± standard deviation. Differences between the groups were analyzed using the Student's t-test and ANOVA. Tukey test was used for post hoc analysis. All statistical analyses were conducted three times using GraphPad Prism 8.0. A p-value <0.05 was considered statistically significant.
Results
NaHS alleviated colitis-induced histological injury and inflammatory response
To investigate the effects of NaHS in colitis rats, we detected histological injury and proinflammatory cytokine levels. Compared to the control group tissues, HE-stained tissues from the UC group demonstrated an obviously higher histological score, with increased inflammation, higher severity of cell infiltrates, more severe destruction of the crypt, and greater extent of cell infiltrates (Fig. 1A, B). Furthermore, these cellular alterations were significantly reversed after NaHS treatment (p < 0.05; Fig. 1A, B). Meanwhile, the ELISA results demonstrated that the levels of proinflammatory cytokines IL-1β (Fig. 1C), IL-6 (Fig. 1D), and TNF-α (Fig. 1E) were meaningfully increased in the DSS-induced UC group in vivo. On the contrary, the cytokines were downregulated compared to those in the control group (p < 0.05). NaHS administration also remarkably reversed the upregulated activity of MPO, a hallmark protein of neutrophil infiltration, in colitis rats (Fig. 1F) compared to DSS-treated rats. The findings indicate that NaHS was able to alleviate inflammatory responses in DSS-induced UC.

NaHS treatment apparently alleviates histological injury and proinflammatory cytokine expression in colitic rats. NaHS alleviated inflammatory responses in the colon of DSS-induced ulcerative colitis rats
Downregulation of CSE and H2S in colitis rats treated with NaHS
In the peripheral system, CSE is critical for H2S synthesis (16). We, therefore, measured the level of CSE in NaHS-treated colitic rats. Immunohistochemical analysis of the colon tissues demonstrated increased CSE expression in UC samples and decreased CSE expression in NaHS-treated UC samples (p < 0.01; Fig. 2A, B). In line with the CSE results, the increased H2S level in the model group decreased with NaHS administration (p < 0.01; Fig. 2C), indicating that the endogenous H2S expression was negatively regulated NaHS. These results prove that NaHS supplementation can regulate endogenous H2S, which can be achieved by inhibiting CSE expression in colonic tissues.
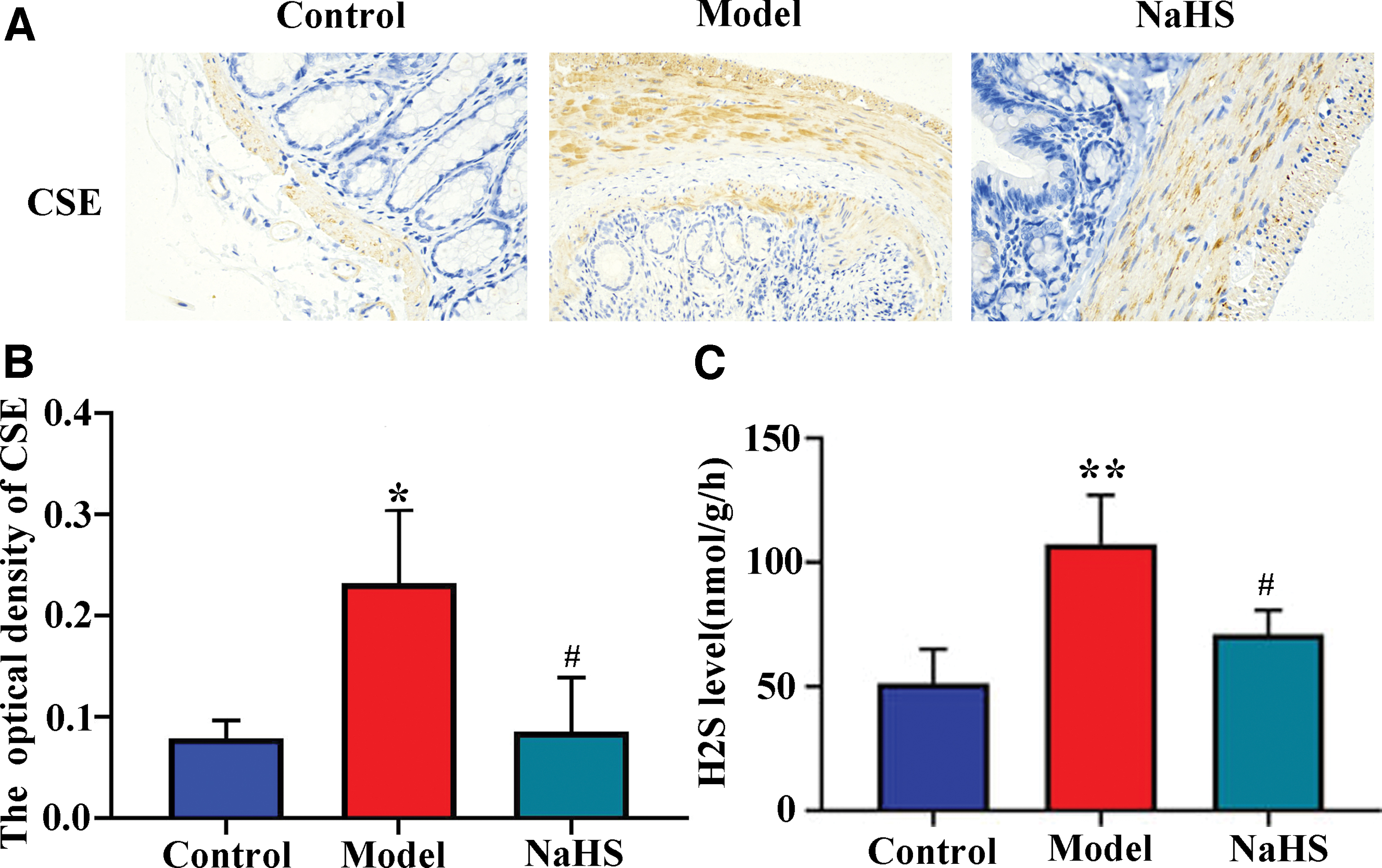
Downregulation of CSE and H2S in colitis rats.
Upregulated LC3B in colitic rats and attenuated ROS level in vitro by NaHS administration
To explore whether autophagy was affected by NaHS administration in LPS-induced inflammation, we analyzed the in vivo expression of LC3B. IF staining demonstrated that the expression of LC3B was significantly decreased in UC compared with control rats, whereas it was upregulated by NaHS (p < 0.05; Fig. 3A, B) in inflamed colonic tissues of UC rats. The results indicate that NaHS treatment can modulate autophagy in vivo. To further determine the role of NaHS in UC, we established a cellular model of colitis using LPS-stimulated H2940 cells in vitro. The H2940 cells were experienced with the LPS procedure in vitro. Intracellular ROS was upregulated in LPS-stimulated cells, which was reversed after treatment with NaHS (p < 0.05; Fig. 3C, D). These findings implicate the protective role of NaHS in colitis-induced oxidative stress.
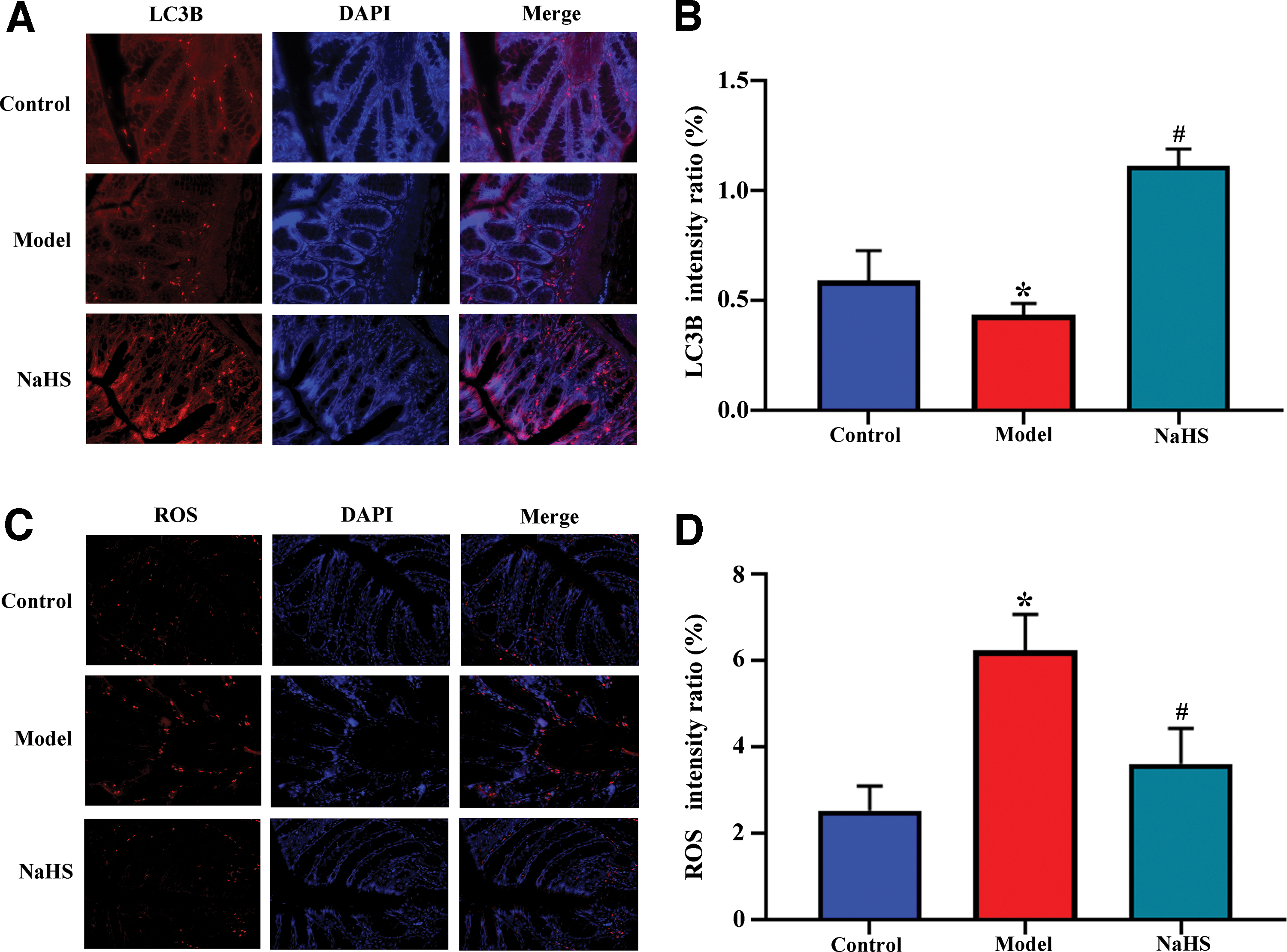
Upregulated microtubule-associated protein 1A/1B-light chain 3B in colitis rats and reduced ROS in LPS-stimulated cells after NaHS treatment.
NaHS reduced CSE and NLRP3 levels in LPS-stimulated cells
We also measured CSE level in vitro in LPS-stimulated H2940 cells. The elevated mRNA expression of CSE was observed in LPS-stimulated cells, whereas it was reduced after NaHS treatment, as was detected by qRT-PCR (p < 0.05; Fig. 4A). We then assessed whether the NLRP3 inflammasome was involved in colon injury and whether inflammatory cytokines were induced during UC. The results from qRT-PCR revealed that the mRNA expression of NLRP3 was remarkably increased in LPS-stimulated cells but was decreased after NaHS administration (p < 0.05; Fig. 4B). The levels of CSE and NLRP3 protein were further confirmed by the western blot, and the data were consistent with those of qRT-PCR (p < 0.05; Fig. 4C, D).
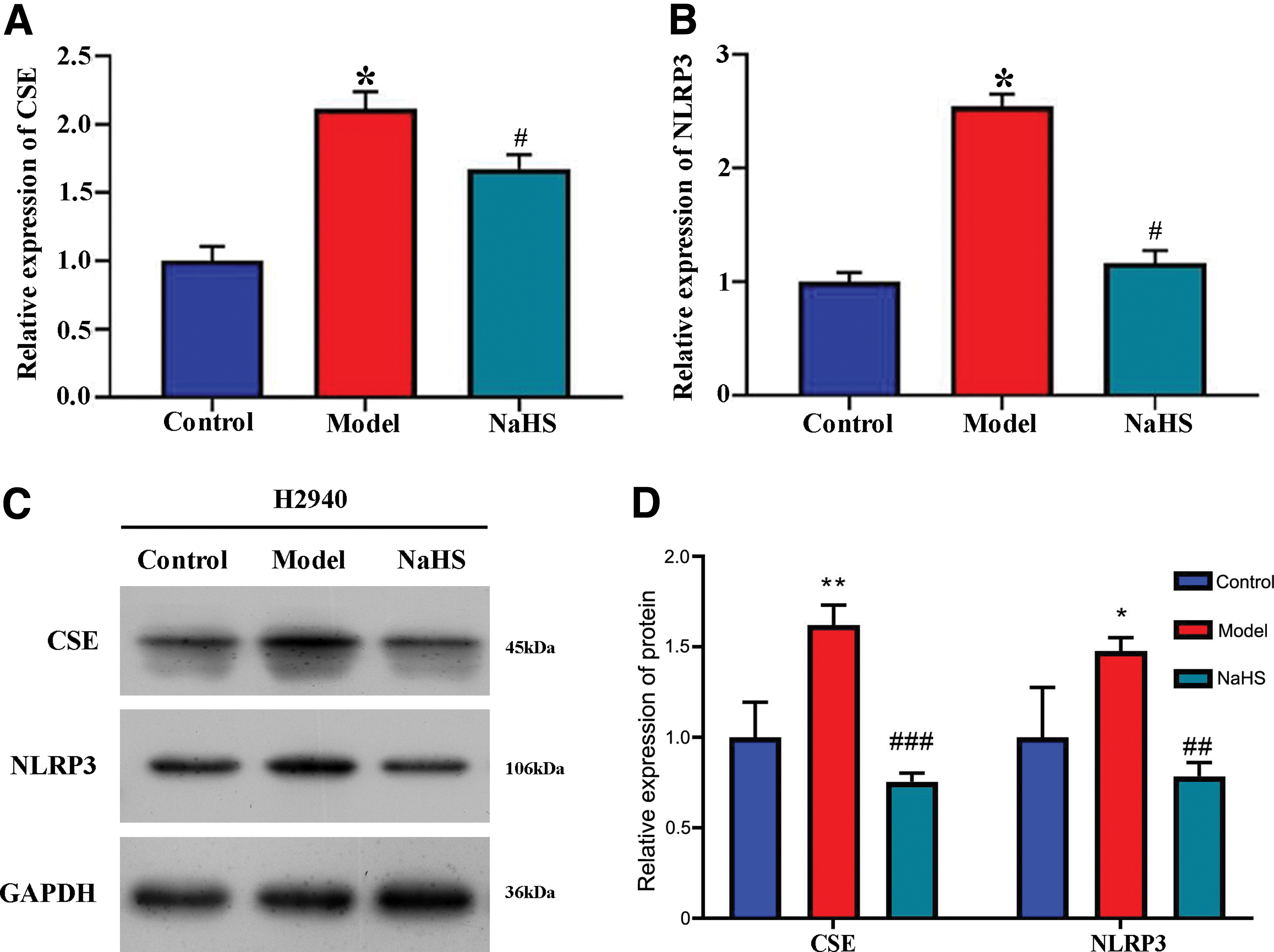
Downregulation of CSE and NLRP3 expression in LPS-stimulated cells after NaHS treatment. The relative expression of
NaHS-enhanced autophagy was determined by regulating the levels of autophagy-related proteins
To further confirm the impact of NaHS on autophagy, the proteins beclin 1 and p62, and the conversion of LC3-I to LC3-II, were detected in vitro using western blot. The LC3-II-to-LC3-I ratio and the level of beclin 1 were significantly elevated, whereas p62 levels were decreased after NaHS administration in LPS-stimulated cells compared with the model group cells (p < 0.05; Fig. 5A, B). Further, LC3 level, as was detected by IF, was noticeably elevated by NaHS in LPS-treated cells compared with the model group cells (p < 0.05; Fig. 5C). Transmission electron microscopy revealed the involvement of NaHS in autophagosome formation in LPS-treated cells (Fig. 5D). These findings indicate that NaHS promotes autophagy of inflammatory H2940 cells.

NaHS promotes the formation of autophagosome in LPS-stimulated cells. Total protein was isolated and submitted to western blot analysis.
A schematic demonstrating the underlying mechanisms of NaHS in colitis
The schematic revealed the underlying mechanisms of NaHS in colitis. Exogenous NaHS alleviated colitis by promoting autophagy and inhibiting the NLRP3 inflammasome (Fig. 6). On the contrary, NaHS suppressed the endogenous CSE-synthesized H2S level and inhibited LPS-induced ROS upregulation, NLRP3 expression, and inflammasome formation, finally alleviating colitis (Fig. 6).

A schematic showing the underlying mechanisms of NaHS on colitis. Color images are available online.
Discussion
Existing research supports the positive effects of NaHS in several diseases. NaHS noticeably alleviated Aβ (1–40)-induced neuroinflammation and apoptosis by suppressing the activities of p38 MAPK and p65 NF-κB (Xuan et al., 2012). Exogenous H2S from NaHS alleviated cardiac injury by reducing apoptosis and inflammation response under oxidative stress (Bai et al., 2018). NaHS enhanced renal function and the histopathological changes of the kidneys, ameliorated LPS-induced inflammation, and oxidative stress, and suppressed NLRP3 expression (Chen et al., 2018). However, data on the role of NaHS in gastrointestinal disorders are limited. In our study, we investigated the potential effects of NaHS treatment on colitis injury both in vivo and in vitro. NaHS treatment considerably reduced histological injury, upregulated LC3B expression, and reduced the expression of proinflammatory cytokines, CSE, and endogenous H2S in colitic rats in vivo. In addition, NaHS remarkably attenuated ROS, CSE, and NLRP3 levels and enhanced autophagy, and promoted autophagosome formation in LPS-stimulated cells in vitro.
Published literature reveals that H2S is mainly generated from Cys or its derivatives by CSE and CBS (Paul and Snyder, 2012). NaHS at 500 μM significantly attenuated Caco-2 monolayer barrier injury and TNF-α/interferon-γ (IFN-γ)-induced p65 NF-kB activation in vitro, highlighting the protective effect of H2S in the barrier function of intestinal epithelial cells (Chen et al., 2015). Several studies have verified the effect of H2S donors on colitis both in vivo and in vitro. Mesalamine (ATB-429), an H2S-releasing derivative, reduced colitis severity, granulocyte infiltration, and mRNA expression of proinflammatory cytokines and chemokines (e.g., TNF-α, IFN-γ) in a trinitrobenzene sulfonic acid-induced colitis mouse model (Fiorucci et al., 2007). GYY4137, a slow-release H2S donor, improved intestinal barrier injury and attenuated inflammatory response in DSS-induced colitis (Zhao et al., 2016). Lawesson's reagent, an H2S donor, decreased the extent of colonic inflammation (Kupai et al., 2018). In our study, we confirmed that NaHS alleviated histological injury and reduced the levels of proinflammatory cytokines IL-1β, TNF-α, and IL-6 in rats with DDS-induced colitis, indicating a therapeutic role of the compound in colitis. In a previous report, NaHS attenuated inflammatory response induced by DSS in mice and decreased the mRNA expressions of IL-1β, IL-17, and TNF-α in Caco-2 cells treated with 2% DSS (Chen and Liu, 2016). These findings, together with our results, confirm the protective effects of NaHS in experimental models of colitis both in vivo and in vitro. Moreover, data from that report indicated that NaHS exerted its effects through NF-κB activation inhibition (Chen and Liu, 2016). In terms of the action mechanisms, we focused on how NaHS regulates the CSE enzymes. Our results revealed that NaHS downregulated the increased CSE level in the colitis group. The decreased expression of CBS, another predominant enzyme in H2S generation in colonic epithelial cells, exacerbated TNF/IFN-induced intestinal barrier injury by augmenting the NF-κB p65-modulated MLCK-P-MLC signaling pathway in Caco-2 monolayers (Chen et al., 2019). In our study, NaHS reversed the upregulated MPO activity induced by colitis, suggesting the role of the neutrophil infiltration marker in the disease.
The NLRP3 inflammasome is implicated in the pathogenesis and progression of IBD. Interactions among the gut homeostasis, mucosal immune response, and NLRP3 inflammasome are complex (Shao et al., 2019; Zhen and Zhang, 2019). In colitic mice, an NLRP3 inflammasome inhibitor was able to reduce colonic inflammation (Perera et al., 2018). In the present study, NaHS treatment remarkably inhibited NLRP3 expression in LPS-stimulated cells. Few studies have demonstrated that bioactive components, such as brusatol and evodiamine, from Chinese medicines, could ameliorate experimental colitis by regulating NF-κB and NLRP3 inflammasome (Zhou et al., 2018; Shen et al., 2019). Therefore, we assume that the protective effect of NaHS in reducing colitis-induced histological injury and proinflammatory cytokine expression is closely related to NLRP3 inflammasome inhibition.
Autophagy is a precisely regulated system modulated by several proteins. Recently, pharmacological autophagy regulators have been proposed to have a therapeutic potential in IBD (Ke et al., 2016; Retnakumar and Muller, 2019). Our study revealed the upregulation of LC3-II-to-LC3-I ratio, the vital marker of autophagy, and autophagy protein beclin 1 and downregulation of p62 after NaHS administration. The increased autophagy after NaHS treatment observed under electron microscope was also utilized for a compensatory mechanism. These data indicate that NaHS activated autophagy and modulated the associated proteins. A previous study demonstrated that autophagy exerted an inhibitory effect on inflammation activation under hypoxic conditions and that NLRP3 expression and NF-κB signaling were suppressed by hypoxia-induced autophagy (Cosin-Roger et al., 2017). Furthermore, NaHS activated liver autophagy through the AMPK-mTOR pathway, ameliorating nonalcoholic fatty liver disease (Sun et al., 2015). In line with these studies, we found that the enhanced autophagy appeared with the inhibited inflammation response after NaHS treatment, suggesting a relationship between NaHS-induced autophagy promotion and inflammation reduction. We supposed that the inhibitory effect of autophagy on inflammation response may be a therapeutic mechanism of NaHS.
In conclusion, our research verified that NaHS administration improves DDC-induced UC in rats. As an exogenous H2S donor, NaHS demonstrated anti-inflammatory effects by inhibiting the expression of inflammatory cytokines and MPO, a neutrophil cell infiltration marker. Importantly, NaHS restored the impaired autophagy in colitic rats in vivo and in LPS-stimulated cells in vitro. As such, administration of NaHS appears to protect the colon against injury and reduce inflammation, implying its potential for treating UC.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (grant number 81660097, 81960106).