
Abstract
The vascular endothelium, which plays an essential role in maintaining the normal shape and function of blood vessels, is a natural barrier between the circulating blood and the vascular wall tissue. The endothelial damage can cause vascular lesions, such as atherosclerosis and restenosis. After the vascular intima injury, the body starts the endothelial repair (re-endothelialization) to inhibit the neointimal hyperplasia. Endothelial progenitor cell is the precursor of endothelial cells and plays an important role in the vascular re-endothelialization. However, re-endothelialization is inevitably affected in vivo and in vitro by factors, which can be divided into two types, namely, promotion and inhibition, and act on different links of the vascular re-endothelialization. This article reviews these factors and related mechanisms.
Introduction
The vascular endothelial cell layer is the physical barrier of arterial vessels. The injury and the dysfunction of the vascular endothelial cells are the initial links of atherosclerosis (AS) and vascular restenosis (RS). The AS is harmful to human health. AS risk factors (such as high fat, hyperglycemia [HG], and hyperhomocysteinemia [HHcy]) can damage the integrity and the function of the vascular endothelium. Several mechanisms in the body repair the damaged vascular endothelium and maintain the integrity of the vascular endothelium. The reaction mechanism, namely, the re-endothelialization of the vascular endothelium, is an important protective mechanism to prevent AS (Bai et al., 2010; Li et al., 2021).
The percutaneous coronary intervention (PCI) can quickly and effectively relieve the symptoms of patients with coronary heart disease, but the in-stent restenosis (ISR) can cause a relatively poor prognosis (Sun et al., 2017). The main cause of the ISR is the exfoliation injury of the vascular intima, which leads to platelet activation, local inflammation, and the proliferation, migration, and secretion of the extracellular matrix of media smooth muscle cell (SMC) layer due to nudity, and furthers the intimal hyperplasia (Chaabane et al., 2013; Wang et al., 2021). Therefore, accelerating the re-endothelialization and the repair of damaged intima can effectively reduce the ISR.
Previous studies have shown that angiogenesis is a process in which new blood vessels are generated from existing blood vessels (Hanahan and Folkman, 1996). Moreover, angiogenesis involves the interaction between endothelial cells and blood cells and SMCs and extracellular matrix components (Tang and Conti, 2004). However, endothelial progenitor cell (EPC) is considered to be one of the cell sources involved in angiogenesis (Rae et al., 2011). It has a high ability of proliferation and differentiation, can differentiate into endothelial cells, and promote vascular re-endothelialization and angiogenesis (Yoder, 2012; Siavashi et al., 2016; Rezabakhsh et al., 2017).
The traditional view is that repair of vascular endothelium after the vascular endothelial injury is made up of vascular endothelial cells that migrate from adjacent vascular endothelial cells to the injured site and vascular endothelial repair be carried out. This process is involved and regulated by a variety of signals. For example, p110γPI3K signaling (Huang et al., 2016), miRNA-126 signaling (Schober et al., 2014), Notch1 signaling (Schober et al., 2014), apelin signaling (Masoud et al., 2020), vascular endothelial growth factor (VEGF) signaling (Hutter et al., 2004; Buchwald et al., 2006), and so on.
Later studies have confirmed, by mobilizing EPC to migrate to the injured site, proliferate and differentiate into endothelial cells, repair damaged vascular endothelium, and promote re-endothelialization. However, the insufficient and the delayed re-endothelialization of the EPC are considered the two main aspects for the formation of AS plaque and RS (Chen et al., 2015). Therefore, the re-endothelialization of EPC should be accelerated to prevent the AS and the RS. However, the re-endothelialization of blood vessels occurs in the body and is inevitably affected by various factors in vivo and in vitro (Li et al., 2012a). This factor can act on the mobilization, homing (adhesion and migration), proliferation, and differentiation of EPC, and related mechanisms are also diverse. This article provides a review of the blood vessel re-endothelialization of EPC.
EPC and Vascular Endothelial Repair
The EPC, the precursor of vascular endothelial cells, can differentiate into endothelial cells in vitro and bind to active angiogenesis sites. This binding significantly promotes the formation of new blood vessels after tissue ischemia in vivo (Asahara et al., 1997).
Subsequent studies show that the EPC plays a therapeutic role in various vascular diseases (Tan et al., 2006; Xu et al., 2012; Mitchell et al., 2017; Yao et al., 2020). Moreover, studies show that in patients with cardiovascular risk factors or cardiovascular diseases (CVDs), the number and the functional activity of EPC decrease (Xu et al., 2012). Although the mechanism of EPC-mediated vascular repair remains unclear, many animal studies show that the circulating EPC may provide an endogenous repair mechanism to combat the endothelial damage induced by persistent risk factors and replace dysfunctional endothelial cells under physiological conditions (Hirsch et al., 1983; Daub et al., 2006; de Boer et al., 2006; Massberg et al., 2006; Stellos et al., 2008; Tanaka et al., 2008; Liu et al., 2009).
However, it was later found that the bone marrow (BM) microenvironment (stem cell niche) is composed of fibroblasts, osteoblasts, and endothelial cells (Zhang et al., 2014). The EPC binds to the vascular cell adhesion molecule 1 (VCAM-1) on the surface of BM stromal cells through integrins (such as α4β1 and β3) (de la Puente et al., 2013). When stromal cells are stimulated by various factors, they can induce the EPC to proliferate and transform into functional cells with regenerative potential, migration, and tissue implantation (Tilling et al., 2009).
The EPC can secrete many angiogenic factors and accelerate the maturation of endothelial cells and the proliferation, differentiation, and migration of other peripheral EPC, thereby repairing the damaged vascular endothelium and promoting the re-endothelialization (Urbich et al., 2005). These studies show that EPC plays an essential role in promoting endothelial repair after endothelial injury.
Re-Endothelialization of the EPC
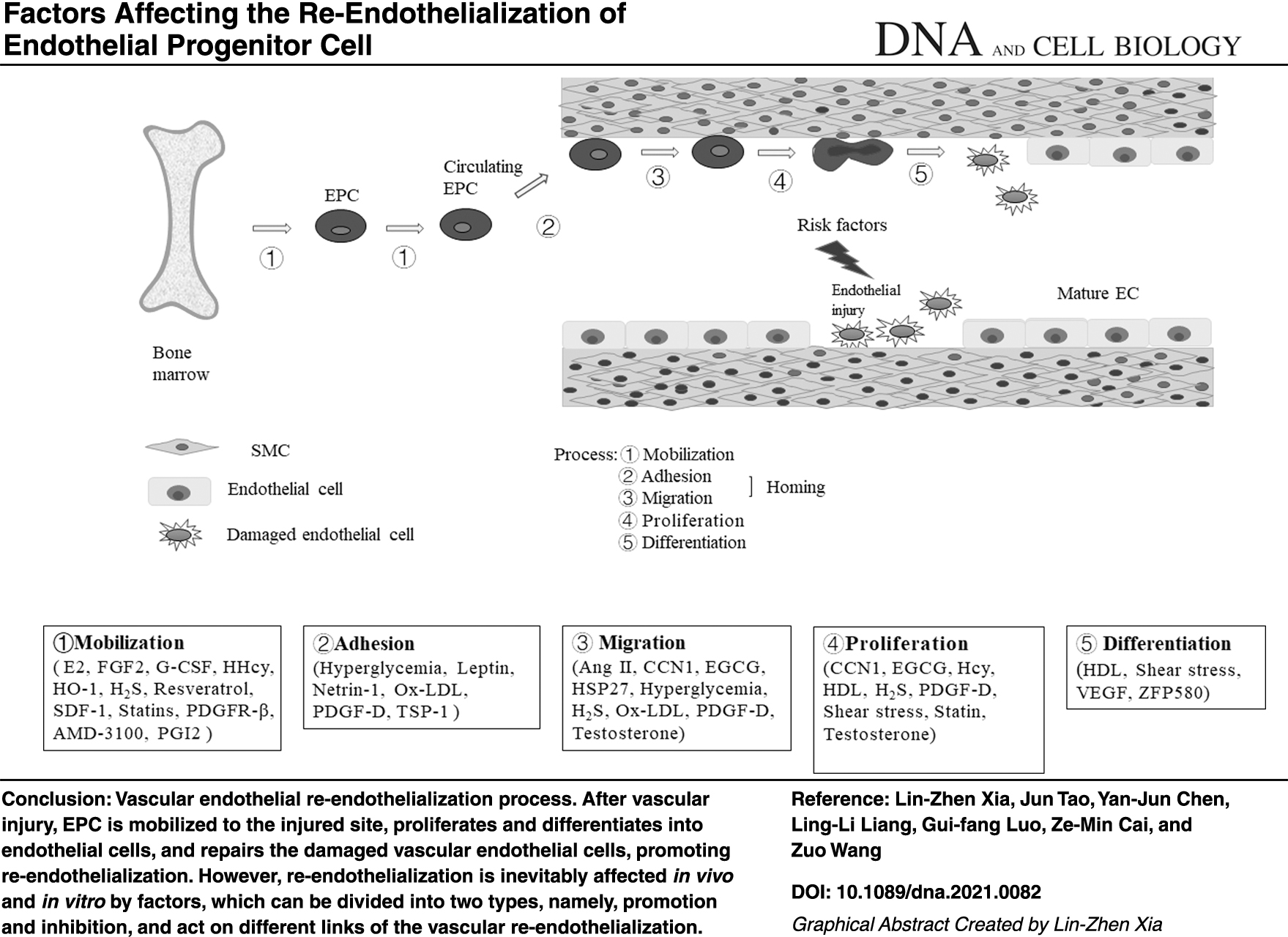
The EPC mediates the re-endothelialization after the vascular injury through multiple steps, such as mobilization, homing (including adhesion and migration), proliferation, and differentiation (Hill et al., 2003; Yeh et al., 2017) (Fig. 1).

Vascular endothelial re-endothelialization process. After vascular injury, EPC is mobilized to the injured site, proliferate and differentiate into endothelial cells, and repair the damaged vascular endothelial cells, which promote re-endothelialization. EPC, endothelial progenitor cell.
The mobilization of EPC is the first step and is subject to strict supervision. EPC resides within the BM and is rarely found in the peripheral blood. When the EPC in the BM is stimulated by various factors, they migrate to the peripheral blood, which is a process called the EPC mobilization (de la Puente et al., 2013). Various cytokines and drugs can promote EPC mobilization (Liu et al., 2013b; Pai et al., 2016). The EPC mobilization is a complex process and can be achieved by a single factor that potentially acts in multiple ways or by a synergistic effect of multiple factors (Sun et al., 2020).
At present, an in-depth research is conducted on the improvement of adult angiogenesis, but knowledge on the mechanism of EPC homing remains lacking (Ferrara et al., 1996; Vajkoczy et al., 2003; Urbich and Dimmeler, 2004). The initial steps of EPC homing to ischemic tissue include the adhesion of EPC to cytokines and ischemia-activated endothelial cells and the migration of EPC to the endothelial cell monolayer (Vajkoczy et al., 2003). Integrins mediate the adhesion of cells to the extracellular matrix and endothelial cells (Springer, 1994; Muller, 2002). Considering that the homing of stem cells in vivo to tissues involves adhesion and migration, 80 integrins, such as β2-integrin and β4–1 integrin, may be involved in the transfer of EPC to ischemic tissues and the homing process (Urbich and Dimmeler, 2004).
The differentiation of EPC into functional endothelial cells is crucial to re-endothelialization and vascular function maintenance. However, the underlying molecular mechanisms, especially the microenvironment of the injured vessel wall that regulates the EPC differentiation and the re-endothelialization during the vascular repair process, remain poorly understood. Various cytokines and drugs can promote EPC differentiation. For example, the VEGF and its receptors play a vital role in stimulating the differentiation of endothelial cells (Ferrara et al., 1996). Pim-1 kinase is a downstream effector of VEGF, VEGF can promote the differentiation of EPC into endothelial cells by acting on Pim-1 kinase (Zippo et al., 2004). Therefore, we are briefly reviewing the mechanism of re-endothelialization mediated by EPC after vascular injury.
The Signaling Pathway Regulating the Re-Endothelialization
It is well known that various mechanisms regulate the vascular re-endothelialization, but the most studied pathways are the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/endothelial nitric oxide synthase (eNOS), the stromal cell-derived factor-1 (SDF-1)/CXCR4/JAK-2, the nuclear factor-kappa B (NF-κB)/survivin, and the NO/cGMP/p38MAPK signal pathways, which are essential for the vascular re-endothelialization and important targets for monitoring and intervention (Fig. 2).

The primary mechanisms regulate vascular re-endothelialization. Various mechanisms regulate the vascular re-endothelialization, but the most studied pathways are the PI3K/Akt/eNOS, the SDF-1/CXCR4/JAK-2, the NF-κB/survivin, and the NO/cGMP/p38MAPK signal pathways, which are essential for the vascular re-endothelialization and essential targets for monitoring and intervention.
The PI3K/Akt/eNOS signaling pathway
The PI3K/Akt/eNOS signaling pathway is an important pathway for endothelial repair after vascular injury (Hu et al., 2015). Previous research found that the eNOS mobilizes the EPC cycle (Wang et al., 2007). It is suggested that eNOS may be involved in the process of endothelial repair promoted by EPC. Later studies have found that the eNOS is a downstream effector of the PI3K/Akt survival signaling pathway, which participates in the PI3K/Akt-mediated endothelial functional changes. Therefore, the activation of the PI3K/Akt signaling pathway induces the eNOS phosphorylation and the nitric oxide (NO) production, advancing the endothelial repair after vascular injury (Everaert et al., 2010; Huang et al., 2010; Wegiel et al., 2010).
For instance, the shear stress (Yang et al., 2012a) and erythropoietin (EPO) (Wu et al., 2019) can accelerate the re-endothelialization of the EPC in injured vessels through PI3K/Akt/eNOS signaling pathway. Moreover, the pitavastatin can facilitate the proliferation of EPC through the PI3K signaling pathway and accelerate the re-endothelialization of the injured site, which was also reported in animal disease models (Liu et al., 2017a). Studies have shown that PI3K, Akt, and eNOS are downstream signaling molecules of epidermal growth factor homology domain 2 (Tie2) (Yang et al., 2012b).
Tie2 is a significant endothelial-specific receptor tyrosine kinase (Yang et al., 2012b). Evidence supports the hypothesis that Tie2 and its ligands, for example, angiopoietin-2 (Ang2), contribute to angiogenesis (Kim et al., 2006). Besides, studies show that aliskiren, an active direct renin inhibitor, can improve the function in both in vitro and in vivo re-endothelialization capability of EPC through the activation of the Tie2/PI3K/Akt/eNOS signaling pathway (Yao et al., 2020). Therefore, the PI3K/Akt/eNOS signaling pathway plays an essential role in promoting vascular re-endothelialization by EPC. In addition, some studies have found that β2AR/PI3K/Akt (Galasso et al., 2013; Hu et al., 2020) signaling pathway is also involved in EPC-mediated re-endothelialization.
The SDF-1/CXCR4/JAK-2 signaling pathway
The SDF-1, a vital stem cell chemokine, is considered to participate in the mobilization and the homing of EPC after the endothelial injury and accelerate the re-endothelialization of the EPC (Yin et al., 2010). The SDF-1 is linked to its CXCR4 ligand through the SDF-1/CXCR4 axis. Then, the EPC is recruited and homing done to the damaged tissue (Noels et al., 2014; Li et al., 2017b). Therefore, the SDF-1/CXCR4 axis is related to EPC mobilization and homing.
The JAK-2 is an important effector molecule downstream of CXCR4 and is involved in CXCR4-mediated cell function changes. Studies have shown that the enhancement of the EPC function by the CXCR4 transfer depends on the JAK-2 signaling pathway mediated by the CXCR4 (Chen et al., 2010). Thus, the SDF-1/CXCR4/JAK-2 signaling pathway may be related to the promotion of endothelial repair after endothelial injury by EPC.
For example, plerixafor (also known as AMD3100) is a specific antagonist of the SDF-1/CXCR4 axis. The AMD3100 can inhibit CXCR4 (Jiang et al., 2020), thereby blocking the contact between the SDF-1 and the CXCR4 and making the role of SDF-1 in the EPC migration blocked, but the AMD3100 can mobilize CD34+ cells, boost EPC to enter the blood, and promote the formation of new endothelial cells. However, the mechanism by which AMD3100 advances the EPC migration is unclear but maybe through competition the SDF-1-/CXCR4 axis enhances the effect of EPC migration, thereby promoting the re-endothelialization (Li et al., 2017c). Therefore, the SDF-1/CXCR4/JAK-2 signaling pathway promotes vascular re-endothelialization by affecting EPC function.
The NF-κB/survivin signaling pathway
NF-κB as the hallmark of tumorigenesis has been proposed to be a crucial regulatory family for inflammation, immunity, and cell survival (Altieri, 2008; Ghosh et al., 2012). Survivin is a new member of the apoptosis inhibitor protein family. NF-κB is the upstream signaling of survivin, which can regulate the expression of survivin (Ghosh et al., 2012; Cui et al., 2017).
To verify the relationship between NF-κB and survivin, it has been studied that NF-κB activator or inhibitor was used to regulate the expression of NF-κB in bladder cancer cell lines. The activation of NF-κB significantly upregulated the expression of survivin. On the contrary, the inhibitor selectively inhibited the phosphorylation of I-kinase (IKK) resulting in the inactivation of NF-κB, thus downregulating the expression of survivin (Cui et al., 2017). And later studies found that the inhibitor of DNA-binding 1 (Id1) can promote angiogenesis by activating the NF-κB/survivin-related signaling pathway (Li et al., 2012b, 2019) and promote the proliferation of EPC in vitro (Li et al., 2012b). Therefore, the NF-κB/survivin signaling pathway plays an important role in promoting vascular re-endothelialization, inhibiting neointimal hyperplasia (NIH), and preventing vascular RS (Li et al., 2019).
The NO/cGMP/p38MAPK signaling pathway
eNOS is known to be a vasodilator with anti-inflammatory and antithrombotic properties. Studies have found that enhanced eNOS activity is correlated with increased numbers of circulating EPC (Thum et al., 2006). NO produced by eNOS is a key regulator of vascular tone and structure (Kataoka et al., 2017; Swelum et al., 2017; Tsikas and Kinzel, 2018), and impaired NO production leads to endothelial dysfunction (Oemar et al., 1998). And some studies have found that testosterone may participate in endothelial repair by regulating the expression and activity of eNOS, increasing the level of NO, and regulating the proliferation and migration of EPC (Hotta et al., 2019). Therefore, the NO produced by eNOS may be related to EPC-mediated endothelial repair.
Otherwise, the study has shown a stimulatory effect of growth hormone (GH) on circulating EPC levels (Thum et al., 2007b); GH improves markers of NO bioavailability and circulating EPC levels. In conclusion, GH may be involved in EPC-mediated endothelial cell repair by improving the bioavailability of NO (Thum et al., 2007a). Moreover, some studies have found that NO produced by eNOS can promote the formation of cGMP and p38MAPK. Some studies have further proved that p38MAPK is located downstream of cGMP vascular SMCs (VSMCs) where migration was abrogated by netrin-1 through a NO/cGMP/p38MAPK signaling pathway, while EPC homing was induced (Liu et al., 2017b). Therefore, the NO/cGMP/p38MAPK signaling pathway may play an important role in EPC-mediated re-endothelialization.
Others
Various mechanisms regulate vascular endothelial re-endothelialization. Other signaling pathways, such as the eNOS/NO, the Sirtuin3 (SIRT3)/superoxide dismutase (SOD)2, and the mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) signaling pathways, can also regulate the vascular re-endothelialization.
Factors Affecting Re-Endothelialization
The re-endothelialization process of EPC is inevitably affected by internal and external factors. These factors can be divided into two types, namely, promotion and inhibition, which can act on the mobilization, homing (adhesion and migration), proliferation, and differentiation of EPC.
Factors inhibiting the re-endothelialization
Relatively few factors inhibit the mobilization, homing (adhesion and migration), proliferation, and differentiation of EPC (Table 1).
Factors That Inhibit Re-Endothelialization of Blood Vessels
Ang II, angiotensin II; DPP-4, dipeptidyl peptidase 4; EPC, endothelial progenitor cell; GSK3β, glycogen synthase kinase 3β; Hcy, homocysteine; HG, hyperglycemia; HHcy, hyperhomocysteinemia; OS, oxidative stress; Ox-LDL, oxidized low-density lipoprotein; VEGF, vascular endothelial growth factor.
Rapamycin
The rapamycin is an immunosuppressant added to a drug-eluting stent that can prevent RS and damage the vascular re-endothelialization (Ying Qian and Feng 2016). The rapamycin inhibits the proliferation and the migration of EPC by inducing the EPC apoptosis and reducing the expression of VEGF, thereby inhibiting the re-endothelialization after the drug-eluting stent implantation (Zaruba et al., 2009). Although its mechanism is not completely clear, it is of great significance in inhibiting the occurrence and development of AS.
Proteins and enzymes
The angiotensin II (Ang II) acts on the Ang II type 1 receptor (AT1-R) (Petoumenos et al., 2009) and participates in the formation of AS. When the vascular endothelium is damaged, the angiotensin can activate the AT1-R, induce oxidative stress (OS), reduce the number of early-growth EPC, impair migration ability of early-growth EPC (Endtmann et al., 2011), and inhibit the re-endothelialization of the damaged vascular endothelium.
The oxidized low-density lipoprotein (ox-LDL) affects the biological activity of EPC by downregulating the eNOS and damages its adhesion, migration, and ability in a dose-dependent manner (Imanishi et al., 2004; Wang et al., 2004; Ma et al., 2006), thereby inhibiting the re-endothelialization of the damaged endothelium (Cheng et al., 2007).
The dipeptidyl peptidase 4 (DPP-4) exists freely in serum and can be expressed on the surface of EPC. The interaction of SDF-1/CXCR4 is regulated by DPP-4, which cleaves two N-terminal amino acids (X-Pro) and inactivates the SDF-1 (Zaruba et al., 2009).
It is suggested that DPP-4 can inhibit EPC-mediated re-endothelialization by the SDF-1/CXCR4 signaling pathway. And some studies have found that inhibiting the activity of DPP-4 can promote endothelial cell regeneration (Brenner et al., 2014). These results suggest that DPP-4 may be involved in the re-endothelialization of EPC through the SDF-1/CXCR4 signaling pathway. The glycogen synthase kinase 3β (GSK3β) is a serine/threonine kinase that can regulate the survival and the function of various transcription factors (Park et al., 2003). Studies show that the GSK3β participates in the regulation of the EPC-mediated re-endothelialization. Although its mechanism is not completely clear, further studies indicate that the inhibition of GSK3β significantly increases the re-endothelialization (Etheridge et al., 2004).
Hyperglycemia
In patients with type 1 (Loomans et al., 2004) and type 2 diabetes (Tepper et al., 2002) and diabetic animals (Schatteman et al., 2000; Tamarat et al., 2004; Kränkel et al., 2005), the number of circulating EPC is reduced, and the functions of EPC, such as adhesion, migration, and paracrine of proangiogenic factors, are impaired, which may be caused by HG. The EPC is increasingly recognized as a potential therapeutic target for the prevention of ischemic vascular disease. HG can change the differentiation direction of BM precursor cells, reduce the generation capacity of angiogenic cells, facilitate the development of pro-inflammatory cells, and inhibit the re-endothelialization of the EPC (Loomans et al., 2009). It is suggested that HG can inhibit re-endothelialization by inhibiting the functions of EPC.
Other
Oxidative stress
The OS refers to the oxidative damage caused by the accumulation of reactive oxygen species (ROS) in vivo or cells due to increased oxygen free radicals or decreased scavenging ability after the body tissues or cells are stimulated by unfavorable factors. Moreover, the OS refers to the oxidative damage process caused by the imbalance of the oxidative and the antioxidant system (Sies, 2015; Li et al., 2020). The OS degrades the vascular re-endothelialization capacity of the EPC by producing ROS and ox-LDL (He et al., 2019). It is indicated that OS can inhibit EPC-mediated re-endothelialization.
Homocysteine and HHcy
The Hcy is an independent risk factor for the AS (Kil et al., 2017). The Cyclin A is a vital regulator of the cell cycle progression at the beginning of DNA replication and mitosis (Kanakkanthara et al., 2016). Studies show that the Hcy may inhibit the proliferation of the EPC by downregulating the expression of the Cyclin A (Zhang et al., 2018) and the inhibition of the vascular re-endothelialization after injury.
The HHcy is considered a significant risk factor for CVD. The HHcy can impair the re-endothelialization ability of endothelial cells by inhibiting the proliferation and the migration of endothelial cells (Tan et al., 2006). Later studies show that the HHcy inhibits the mobilization and the homing of BM-derived EPC to the damaged vascular endothelium and the re-endothelialization after vascular injury (Nelson et al., 2015).
Factors that accelerate the re-endothelialization
Many factors facilitate the mobilization, homing (adhesion and migration), proliferation, and differentiation of EPC, thereby accelerating the re-endothelialization (Table 2).
Factors That Promote Re-Endothelialization of Blood Vessels
AMD-3100, plerixafor; CCN1, rich in cysteine 61; EGCG, epigallocatechin gallate; EPC, endothelial progenitor cell; EPO, erythropoietin; E2, estradiol; GBE, ginkgo biloba extract; G-CSF, granulocyte colony-stimulating factor; GH, growth hormone; GLP-1, glucagon-like peptide-1; HDL, high-density lipoprotein; HSP27, heat-shock protein 27; HO-1, heme oxygenase-1; Id1, inhibitor of DNA-binding 1; KLFs, krüppel-like factors; PDGF-D, platelet derived growth factor D; PDGFR-β, platelet derived growth factor receptor β; PGI2, prostaglandin I2; PNS, panax notoginseng saponins; PPAR-γ, peroxisome proliferator-activated receptor γ; ZFP580, novel zinc finger transcription factor.
Regulation effect of the shear stress on the EPC re-endothelialization
The shear stress is the frictional force of blood flow acting on the surface of endothelial cells and has a regulatory effect on the EPC re-endothelialization of blood vessels (Yamamoto et al., 2003; Kutikhin et al., 2018; Souilhol et al., 2020).
Tie2 is an endothelium-specific receptor tyrosine kinase. The activation of the Ang2/Tie2 signaling pathway can facilitate EPC mobilization, migration, and cell survival (Kim et al., 2006; Shyu, 2006). However, the shear stress can mediate the guanosine triphosphate clohydrolase (GTPCH)/tetrahydrobiopterin (BH4) pathway to ameliorate the decrease in re-endothelialization caused by hypertension-related EPC (Duda et al., 2004; Crabtree et al., 2009; Bai et al., 2017). In addition, the shear stress can directly accelerate the re-endothelialization of the EPC in injured vessels by acting on multiple signaling pathways, for example, the Tie2/PI3K/Akt (Yang et al., 2012a), the β2AR/PI3K/Akt (Galasso et al., 2013; Hu et al., 2020), and the SDF-1/CXCR4/JAK-2 (Xia et al., 2012) signaling pathway.
Hormones
The estradiol (E2) can facilitate the EPC re-endothelialization by acting on the estrogen receptor α (Farhat et al., 1996; Brouchet et al., 2001), further the mobilization of BM-derived EPC, and strengthen eNOS to produce NO (Iwakura et al., 2003). Moreover, testosterone may regulate the expression and activity of eNOS, increase the level of NO, regulate the proliferation and migration of EPC, and participate in re-endothelialization (Hotta et al., 2019). The deficiency of GH is closely related to CVD (Böger et al., 1996; Capaldo et al., 2001). Studies have shown a stimulatory effect of GH on circulating EPC levels (Thum et al., 2007b). GH improves markers of NO bioavailability and circulating EPC levels.
In conclusion, GH may be involved in EPC-mediated endothelial cell repair by enhancing the bioavailability of NO. The prostaglandin I2 (PGI2) is an effective antiatherosclerotic lipid medium, which is produced by vascular endothelial cells. The PGI2 can play various roles by binding to receptors, such as inducing vasodilation, inhibiting the proliferation of VSMCs, and effectively inhibiting platelet activation (Narumiya et al., 1999). Studies show that the PGI2 is necessary for the EPC to complete its function and facilitate re-endothelialization (Kawabe et al., 2010).
The glucagon-like peptide-1 (GLP-1) is secreted by intestinal endocrine cells. The GLP-1 can improve the biological function of EPC by promoting the production of VEGF (Kalka et al., 2000; Young et al., 2002) and accelerate re-endothelialization (Xiao-Yun et al., 2011).
The leptin is a hormone secreted by adipose tissues. The leptin can upregulate the expression of synthase and regulate the adhesion characteristics and the homing potential of EPC, thereby enhancing its ability to facilitate vascular endothelial repair (Schroeter et al., 2008). To sum up, a number of hormones are involved in EPC-mediated re-endothelialization.
Exosomes
Exosomes are small membrane particles (40–100 nm in diameter) originating from the multivesicular bodies of cells and an important part of paracrine (Raposo and Stoorvogel, 2013; De Jong et al., 2014). Studies have found that the EPC can produce exosomes (Chen et al., 2013) and that the exosomes from human umbilical vein endothelial cells (HUVECs) and EPC have similar morphology, size distribution, and characteristics. However, the exosomes from EPC have more abundant biological activity (Kong et al., 2018a; Qu et al., 2020) than those from HUVECs. Therefore, the EPC may be a stable source of exosomes promoting vascular endothelial repair (Hu et al., 2019a, 2019b).
During the RS after angioplasty, exosomes can inhibit NIH in rats by activating the ERK1/2 signaling pathway (Liu et al., 2020), thereby inhibiting the RS of injured blood vessels. Moreover, the protective effect of exosomes may be manifested by promoting endothelial cell repair rather than directly inhibiting the proliferation and the migration of SMCs (Li et al., 2016; Kong et al., 2018a). For example, EPC-derived exosomes have been shown to promote vascular endothelial cell repair by shuttling the miR-21-5p for the regulation of the platelet reactive protein-1 expression (Hu et al., 2019b) (Fig. 3). These studies suggest that exosomes can participate in EPC-mediated vascular re-endothelialization in a variety of ways.

Exosomes promote vascular endothelial cell repair. During the RS after angioplasty, path one (blue arrow): exosomes can inhibit NIH in rats by activating the ERK1/2 signaling pathway. Path two (black arrow): exosomes have been shown to promote vascular endothelial cell repair by shuttling the miR-21-5p for the regulation of the platelet reactive protein-1 expression. NIH, neointimal hyperplasia; RS, restenosis.
Compounds and drugs
As a powerful antioxidant, the epigallocatechin gallate plays an important role in maintaining the endothelium function under diabetes conditions (Suganya et al., 2016). The promotion of the proliferation and the migration of the EPC through the activation of the Akt/eNOS pathway accelerates the re-endothelialization of diabetic rabbits after the carotid artery injury (Huang et al., 2018).
The resveratrol can promote the re-endothelialization by upregulating the eNOS expression and mobilize the BM-derived EPC, inhibit intimal hyperplasia, and facilitate the re-endothelialization of the damaged arterial endothelium (Wang et al., 2007).
Panax notoginseng saponins (PNS) can accelerate the EPC mobilization, upregulate the interaction of SDF-1 and CXCR4, reduce the size of atherosclerotic plaques, and upregulate the vascular endothelium differentiation (Liu et al., 2013a).
Statins (Remm et al., 2018) can accelerate the re-endothelialization by mobilizing the EPC to the site of initial revascularization (Sun et al., 2008; Yu et al., 2019; Chu et al., 2020). For example, the rosuvastatin can mobilize BM-derived EPC, increase the number of circulating EPC, accelerate re-endothelialization, and significantly reduce neointimal formation (Walter et al., 2002; Werner et al., 2002). The pitavastatin can facilitate the proliferation of EPC after carotid artery injury in rats through the PI3K signaling pathway and accelerate the re-endothelialization of the injured site (Liu et al., 2017a). The sitagliptin can accelerate the re-endothelialization of EPC after aneurysmal neck injury through the SDF-1/CXCR4/NRF2 signaling pathway (Brouchet et al., 2001).
Therefore, we know that a variety of compounds and drugs can promote vascular re-endothelialization mediated by EPC in various ways.
H2S
H2S is a new type of gas signaling molecule that can regulate the pathogenesis of various vascular diseases (Yang et al., 2008; Wang et al., 2009), stimulate the proliferation and the migration of endothelial cells, and advance angiogenesis (Cai et al., 2007). Recently, H2S is found to directly facilitate the mobilization of endothelial eNOS-dependent EPC or indirectly trigger VEGF-induced Ca2+ signaling in endothelial cells, enhance the migration, adhesion, and proliferation of EPC in vitro, and contribute to the re-endothelialization of damaged arteries (Ke et al., 2017; Hu et al., 2019c). It is suggested that H2S plays an important role in the process of vascular re-endothelialization mediated by EPC.
Proteins and enzymes
Proteins and enzymes can accelerate re-endothelialization by affecting the homing, proliferation, migration, and differentiation of EPC. The EPO, heme oxygenase-1 (HO-1), granulocyte colony-stimulating factor (G-CSF), and BM fibroblast growth factor-2 (FGF2) facilitate the mobilization of EPC.
The EPO stimulates the proliferation and the differentiation of erythroid progenitor cells (Anagnostou et al., 1994), which significantly inhibits the neointimal formation and promotes endothelial formation by upregulating the expression of EPO receptors and mobilizing the EPC to activate the Akt/eNOS phosphorylation and the NO synthesis (Urao et al., 2006); therefore, EPO can mobilize EPC to promote re-endothelialization. The HO-1 is a stress-inducing enzyme (Abraham and Kappas, 2008). The HO-1 and its by-product (i.e., CO) can repair damaged blood vessels in mice by promoting the mobilization of the EPC. Further experiments show that the mobilization and the re-endothelialization of EPC in mice with HO-1 deficiency are significantly reduced (Lin et al., 2009). Therefore, the HO-1 and CO play an important role in vascular repair by promoting the mobilization of the EPC.
Studies show that the ginkgo biloba extract (GBE), as an exogenous activator of HO-1 (Biddlestone et al., 2007), facilitates the vascular endothelial repair by activating the EPC PI3K/Akt/eNOS signaling pathway and inducing the expression HO-1 of EPC (Wu et al., 2019) through the p38 signaling pathway.
The G-CSF can stimulate the migration of EPC from the BM to the peripheral blood circulation, and advancing the mobilization of EPC can facilitate the early re-endothelialization after the vascular injury and inhibit the formation of neointima (Kong et al., 2004; Yoshioka et al., 2006). Studies show that FGF2 (Cho et al., 2003) can promote re-endothelialization and inhibit the NIH by mediating the E2 to facilitate the EPC mobilization (Fontaine et al., 2006). Therefore, it is found that EPO, HO-1, G-CSF, and FGF2 can facilitate re-endothelialization by mediating EPC mobilization.
Netrin-1, GPVI-CD133, and FOXC2 accelerate the EPC homing. Netrin-1, an axon guide protein (Kennedy et al., 1994), facilitates angiogenesis by producing NO (Nguyen and Cai, 2006; Bouhidel et al., 2014), which inhibits the VSMC migration through the NO/cGMP/p38MAPK pathway, induces homing of EPC after injury, promotes re-endothelialization, and fights against the RS after endothelial injury (Liu et al., 2017b). The GPVI-CD133 is a bifunctional protein that mediates the EPC homing to vascular diseases and facilitates the re-endothelialization of damaged vascular endothelial cells (Langer et al., 2010). The FOXC2 protein is a member of the family of Forkhead/Fox transcription factors (Xue et al., 2008). Studies show that the overexpression of FOXC2 can significantly increase the expression of CXCR4 on the surface of EPC and enhance the homing ability of EPC, thereby improving the benefit of EPC-mediated therapy after vascular injury (Li et al., 2011). Therefore, it is found that Netrin-1, GPVI-CD133, and FOXC2 can facilitate re-endothelialization by mediating EPC homing.
Several factors are involved in promoting the proliferation and differentiation of EPC, including high-density lipoprotein (HDL), heat-shock protein 27 (HSP27), rich in cysteine 61 (CYR61, CCN1), mitochondrial SIRT3, DNA-binding protein 1, and other transcription factors (e.g., Id1), krüppel-like factor (KLF) 10, novel zinc finger transcription factor (ZFP580), platelet-derived growth factor (PDGF)-D, PDGF receptor (PDGFR) β, etc.
The level of HDL is negatively correlated with cardiovascular events and has important vascular protection (Barter et al., 2007). The HDL can facilitate the EPC proliferation and differentiation, upregulate the eNOS and the endothelium-dependent vasodilation, and stimulate the endothelium cell proliferation. Migration promotes endothelial repair (Petoumenos et al., 2009; He et al., 2018). For example, the apolipoprotein-A1 (apo-A1) can facilitate the proliferation of human EPC through the cell surface ATP synthase (González-Pecchi et al., 2015).
The HSP27 is a member of the small heat-shock protein family and has atherosclerotic protective effects. The HSP27 can significantly upregulate the expression and the secretion of the VEGF in the EPC and increase the differentiation of EPC in vitro, which can promote re-endothelialization (Ma et al., 2014). The CCN1 is a secretory stromal cell protein and belongs to the CCN family (Yu et al., 2010). The high expression of the exogenous CCN1 can induce the secretion of VEGF and monocyte chemoattractant protein-1 (MCP-1), facilitate the proliferation of the EPC, and advance the re-endothelialization (Yu et al., 2008). The mitochondrial SIRT3 is a mitochondrial protein deacetylase dependent on the nicotinamide adenine dinucleotide (NAD+). The SIRT3 inhibits the mitochondrial oxidative damage by improving the re-endothelialization ability of EPC by promoting the deacetylation of SOD in hypertension (He et al., 2019).
Id1 is a member of the transcription factor family, which regulates angiogenesis (Sun et al., 1991; Li et al., 2019). The Id1 regulates the number and the biological function of the EPC (Wikström et al., 2006) and advances the re-endothelialization of injured vessels through the NF-κB/survivin signaling pathway (Li et al., 2019). KLFs and the ZFP580 are members of the zinc finger transcription factor family, which coordinates the repair mechanism of vascular endothelial cells and promotes the re-endothelialization after the carotid artery injury (Wara et al., 2013). The ZFP580 affects the antiapoptosis after the myocardial injury (Meng et al., 2014), which increases the expression of the eNOS and the utilization of NO and accelerates the differentiation of the eNOS into endothelial cells through the eNOS/NO signaling pathway (Wei et al., 2015).
The PDGF-D is a member of the PDGF family (Bergsten et al., 2001; Li and Eriksson, 2003). The PDGF-D enhances the angiogenic ability of the EPC by acting on PDGF receptors and helps the EPC proliferation, thereby promoting endothelial repair (Zhang et al., 2019). However, the PDGFR-β can modify and mobilize the EPC, thereby advancing the endothelial repair (Wang et al., 2014). Therefore, it is found that HDL, HSP27, CCN1, mitochondrial SIRT3, Id1, KLF10, ZFP580, PDGF-D, and PDGFR-β can facilitate re-endothelialization by a variety of mechanisms. In addition, the SDF-1, VEGF, and peroxisome proliferator-activated receptor (PPAR)-γ affect the EPC to promote endothelial differentiation in other ways.
It has been mentioned in the mechanism part of this article that SDF-1 is an important stem cell chemokine, which can bind with CXCR4 ligand, participate in the mobilization and homing of EPCs after endothelial injury through the SDF-1/CXCR4/JAK-2 signaling pathway, and promote the re-endothelialization of endothelial cells (Yin et al., 2010).
The VEGF plays an important role in the EPC differentiation and vascular repair (Kalka et al., 2000; Young et al., 2002; Kong et al., 2018b), which involve the MAPK/ERK signaling pathways (Kawasaki et al., 2008; Xu et al., 2008). The VEGF-pretreated EPC transplantation can further the EPC homing and the re-endothelialization and inhibit the neointimal formation. However, these effects can be inhibited by the siRNA (Li et al., 2017a) of Cx43 (Ke et al., 2013; Pfenniger et al., 2013). Therefore, the VEGF can accelerate the EPC differentiation and the vascular endothelial repair through the Cx43, and the repair mechanism may be related to paracrine (Zhang and Huang, 2020).
The PPARα is a crucial regulator of lipid metabolism (Vergori et al., 2015). The PPAR-γ agonist rosiglitazone can reduce the NADPH oxidase of the EPC activity, restore the bioavailability of NO, and improve the ability of re-endothelialization in vivo (Sorrentino et al., 2007).
Summary and Outlook
The vascular endothelial damage is considered the initiation of AS and stenosis. The PCI can benefit CVDs and cause irreversible mechanical damage to vascular endothelial cells, eventually leading to RS. Therefore, it is very important to realize the rapid re-endothelialization of vascular endothelial cells and restore the integrity of vascular endothelial cells and function. Factors that stimulate the re-endothelialization can prevent the RS after interventional surgery and improve vascular homeostasis.
In addition to the factors discussed and summarized in this article, many other factors affect blood vessels, and the factors of re-endothelialization are yet to be discovered. The molecular mechanisms of the vascular re-endothelialization remain limited, and the most studied mechanisms are the PI3K/Akt/eNOS, the SDF-1/CXCR4/JAK-2, the NF-κB/survivin, and the NO/cGMP/p38MAPK signaling pathways. These pathways are critical to re-endothelialization. Many molecular mechanisms are yet to be discovered, and further in-depth understanding is needed.
The relationship between the EPC and the endothelial repair needs experimental demonstration. The fate of endothelial cells during endothelial injury caused by persistent risk factors is unclear. The research on the influence of the re-endothelialization of vascular endothelial cells and the discussion of its mechanism will help us discover potential targets for regulating the re-endothelialization of vascular endothelial cells and will be helpful for the prevention of CVD and the prevention and treatment of AS and RS.
Footnotes
Authors' Contribution
Z.W. conceptualized and designed the original idea. L.-Z.X., J.T. performed a literature search and analysis and wrote the first draft. Y.-J. C., L.-L. L. provided valuable feedback and critically revised the work. L.-Z.X. generated the figures and tables and revised the draft. Supervision was provided by G.-F.L., Z.-M.C., Z.W. All authors read and approved the final article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Natural Science Foundation of China (No.190GZk038).