
Abstract
Papillary thyroid cancer (PTC) is one of the most prevalent endocrine malignancies. Herein, we aimed to provide a new viewpoint for the PTC progression and explore a new target for the effective therapy for PTC. We found that E26 transformation specific (ETS) variant 4 (ETV4, an ETS family transcription factor) was upregulated in PTC tissues and cells. In vitro experiments exhibited that silencing ETV4 suppressed PTC cell proliferation and cell cycle progression, while the overexpression of ETV4 gained the opposite results. Dual-luciferase reporter assay highlighted that ETV4 could upregulate the solute carrier family 7 member 11 (SLC7A11, a key role for cysteine uptake in ferroptosis) transcription by binding to its promoter region directly. Moreover, the viability inhibition of PTC cells induced by the knockdown of ETV4 was at least partly through the promotion of ferroptosis upon the downregulation of SLC7A11. In in vivo experiment, the results showed that the downregulation of ETV4 repressed the tumor development through the low expression of SLC7A11, and the ETV4 overexpression obtained the contrary effects. Overall, the data suggested that the knockdown of ETV4 suppressed the PTC progression by promoting ferroptosis upon SLC7A11 downregulation.
Introduction
Papillary thyroid cancer (PTC) is one of the most prevalent endocrine malignancies, which accounts for ∼80% of all thyroid cancer cases (Abdullah et al., 2019). The exponential incidence of PTC, accompanied by mostly excellent prognosis, leads to a great prevalent population of PTC patients (Kitahara and Sosa, 2016). Despite the mostly good long-term survival, the biological characteristics of PTC are quite diverse, ranging from less aggressive property to metastatic tumors (Tiedje and Fagin, 2020). The treatment for advanced metastatic PTC has remained challenging. About 30% of patients with advanced aggressive PTC could be successfully treated with radioiodine therapy, while the rest of them are resistant to repeat radioiodine activities (McLeod et al., 2019). And the overall 10-year survival rate drops from 92% of the former to 19% of the latter (McLeod et al., 2019). Two-thirds of the patients might be suggested to have a therapy changing from radioiodine therapy to tyrosine kinase inhibitor drugs, which often lead to adverse events from Grade I (weak side effect) to Grade IV (lethal toxicities) (Nguyen et al., 2015; McLeod et al., 2019). Thus, it is essential to explore the pathogenesis of PTC and identify new effective therapeutic strategies for PTC patients.
E26 transformation-specific (ETS) variant 4 (ETV4) is an ETS family transcription factor, which controls the morphogenesis of epithelial organs and regulates the cellular processes, including proliferation, migration, invasion, and apoptosis in various cancers (Oh et al., 2012; Qi et al., 2020). The overexpression of ETV4 has been reported to promote cell proliferation and migration in lung cancer by upregulating the transcription of paxillin and matrix metalloproteinase 1 (MMP1) (Wang et al., 2020). The knockdown of ETV4 has been proven to weaken the proliferation and induce the apoptosis of gastric cancer cells (Zhang et al., 2020). Pieces of evidence have shown that ETV4 may facilitate the PTC tumorigenesis and is linked with the poor prognosis in PTC (Song et al., 2019; Yu et al., 2020). The effects and regulatory mechanism of ETV4 on malignant phenotypes in PTC have evoked substantial attention.
Solute carrier family 7 member 11 (SLC7A11), a major component of cystine/glutamate antiport system XC −, exerts cystine uptake function for glutathione (GSH) biosynthesis and antioxidant defense (Liu et al., 2019). Scholars have discovered that a compound named Erastin interferes with the process of SLC7A11-mediated cystine uptake by binding to solute carrier family 7 member 5 (SLC7A5, an amino acid transporter in XC − system like SLC7A11), which triggers a new form of cell death termed ferroptosis (Koppula et al., 2020). Ferroptosis is defined as an iron-dependent form of programmed cell death driven by cystine depletion, reactive oxygen species (ROS) accumulation, or iron-dependent lipid peroxidation (Mou et al., 2019). Ferroptosis can be suppressed by SLC7A11 through cysteine uptake, GSH synthesis facilitation, and the lipid ROS detoxification promotion (Koppula et al., 2020). Moreover, ferroptosis plays an essential role in metabolism, redox biology, especially in cancer therapy as a potential target (Stockwell et al., 2017). Roh et al. (2016) have found that ferroptotic cell death induced by downregulated SLC7A11 makes cisplatin-resistant cells more sensitive to treatment in head and neck cancer. Weiland et al. (2019) have proven that ferroptosis is associated with the treatment and prevention of brain diseases. In the treatment of advanced liver cancer, the effects of systemic therapeutic drug sorafenib are proven to depend on metallothionein-1G induced ferroptosis suppression, which is also a system therapeutic drug for PTC (Fallahi et al., 2013; Sun et al., 2016). Therefore, we speculated that ferroptosis might play a role in the progression of PTC.
In this work, we aimed to provide a new viewpoint for the PTC pathogenesis and explore a new target for effective therapy for PTC. We found that ETV4 was highly expressed in PTC tissues and cells. The downregulated ETV4 was verified to repress the PTC progression through ferroptosis promotion upon downregulating the SLC7A11 expression. These results revealed that ETV4 might be a potential therapeutic target through ferroptosis regulation in PTC.
Materials and Methods
Ethical statement and patient tissues
The current research was approved by the Shengjing Hospital of China Medical University (approval no. 2019PS322K). Thirty pairs of PTC tissues and normal adjacent tissues were obtained from patients with thyroidectomy in Shengjing Hospital. Written informed consent was obtained from each patient in this research. Tissues were used for further molecular assays. The research involving patient tissues followed the Declaration of Helsinki, and mice xenograft experimental protocols followed The Guideline for the Care and Use of Laboratory Animals. Animal experiment protocols were approved by Shengjing Hospital of China Medical University (approval no. 2019PS296K).
Cell lines
TPC-1 cells were purchased from Procell Life Science & Technology Co., Ltd (Wuhan, China). Nthy-ori 3-1 and IHH-4 cells were purchased from Cobioer Biosciences Co., Ltd (Nanjing, China). GLAG-66 cells were purchased from Cellcook Biotech Co., Ltd (Guangzhou, China). Nthy-ori 3-1 and TPC-1 cells were cultured in RPMI-1640 medium (Sigma, St. Louis, MO) within 10% fetal bovine serum (FBS; Sigma). GLAG-66 cells were cultured in DMEM (Sigma) within 10% FBS. IHH-4 cells were cultured in medium mixed with DMEM and RPMI-1640 (1:1) within 10% FBS. All cells were cultured at a 37°C incubator with 5% CO2. Furthermore, all cells were authenticated by cell line short tandem repeat profiling, and cells were not contaminated by other cell lines or by mycoplasma.
Transfection
For vector transfection, cells were plated into six-well plates and were cultured until reaching 80% confluence. In the in vitro experiments, transiently transfected PTC cells were used for the investigation of ETV4 function (except for clone formation assay). For ETV4 overexpression in GLAG-66 cells, the coding sequence of ETV4 transcript 1 (NM_001986.4) was subcloned into pcDNA3.1 (GenScript) and named ETV4-OE. Next, 2 μg ETV4-OE was transfected into GLAG-66 cells using 6 μL Lipofectamine 2000. For ETV4 knockdown in TPC-1 cells, 100 pmol short interfering RNAs (siRNAs) were synthesized by GenScript (Nanjing, China) and transfected into TPC-1 cells using 6 μL Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. Moreover, for the downregulation of SLC7A11, SLC7A11 siRNAs were synthesized and cotransfected with ETV4-OE into GLAG-66 cells by Lipofectamine 2000. All transiently transfected cells were used for subsequent experiments after 48 h. The siRNA sequences are shown in Table 1.
Synthetic Short Interfering RNAs or Short Hairpin RNA Sequences
ETS, E26 transformation specific; ETV4, ETS variant 4; NC, negative control; shRNA, short hairpin RNA; siRNAs, short interfering RNAs; SLC7A11, solute carrier family 7 member 11.
Stable transfected PTC cells were used for the establishment of xenografts in vivo and the clone assay formation in vitro. pRNAH1.1 (GenScript) was digested with BamHI and HindIII. ETV4 short hairpin RNA (shRNA) was designed with BamHI (at its 5′ end) and HindIII (at its 3′ end) as cloning sites and diluted with 50 μL annealing buffer at 95°C for 5 min. After being cooled down to room temperature, shRNAs were inserted into pRNAH1.1 using T4 DNA ligase at 16°C overnight. The recombinant plasmid was then transformed into E. coli DH5α with calcium chloride method. Six colonies were picked, and then plasmid DNA was prepared. Subsequently, the plasmid DNA was identified through restriction digest and sequence analyzing. Then, recombinant plasmid and negative control (NC) plasmid were transfected into TPC-1 cells. The shRNA sequences are shown in Table 1. These stable transfected GLAG-66 cells or TPC-1 cells were further screened by 300 μg/mL G418.
Inhibitor or inducer treatment
TPC-1 cells transfected with ETV4 siRNA were incubated with 0.5 μM ferrostatin-1 (Fer-1, a ferroptosis inhibitor; MedChemExpress, Shanghai, China) or 10 μM Z-VAD-FMK (ZVAD, an apoptosis inhibitor; MedChemExpress) for 24 h, respectively. GLAG-66 cells transfected with ETV4-OE were incubated with 20 μM Erastin (a ferroptosis inducer; MedChemExpress) for 24 h.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from PTC cancer tissues or transfected cells using TRIpure (BioTek Instruments, Winooski, VT), respectively. First strand complementary DNA was synthesized using Super M-MLV reverse transcriptase (BioTek Instruments). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using 2 × Taq PCR MasterMix (BioTek Instruments) in the presence of SYBR Green (Sigma). The 2−ΔΔCT method was utilized for quantifying the gene expression. Primers were synthesized by GenScript, and the sequences are shown in Table 2.
The Primer Sequences of Quantitative Real-Time Polymerase Chain Reaction
Western blot
Total protein was isolated from the transfected cells or the xenograft tumors, respectively. The protein concentration was detected by BCA Protein Assay Kit (Beyotime, Shanghai, China). The protein was loaded onto a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA). The bands were incubated with primary antibodies ETV4 (A5797, 1:1000; ABclonal, Wuhan, China), SLC7A11 (A2413, 1:400; ABclonal), or β-actin (sc-47778, 1:1000; Santa Cruz, TX) at 4°C overnight after being blocked with 5% skimmed milk. Next, the strips were incubated with goat anti-mouse or goat anti-rabbit HRP-conjugated secondary antibodies (A0208 or A0216, 1:5000; Beyotime) at 37°C for 45 min. The probes were photographed with an ECL reagent (Beyotime). The target protein expression was analyzed by Gel-Pro-Analyzer (Beijing Liuyi Biotechnology, China).
Colony formation assay
Three hundred transfected cells were planted into 35 mm cell culture dish at 37°C with 5% CO2. Colonies were formed ∼2 weeks later. Next, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, followed by rinsing with PBS twice. Cells were further stained with Wright-Giemsa Stain Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) for 5 min. Images were captured by IXB3 microscope (Olympus, Tokyo, Japan). The colony formation rate was calculated as follows: (the count of colonies/the count of cells that were planted) * 100%.
Cell Counting Kit-8 assay
The ability of cell proliferation was detected by the Cell Counting Kit-8 (CCK-8; Beyotime). 4 × 103 cells per well were plated into 96-well plates and were cultured for 0, 12, 24, 48, 72, or 96 h after transfection. A 10 μL CCK-8 was added into each well for 1 h. The absorbance was measured at 450 nm.
The cell viability of PTC cells was evaluated by the CCK-8. 4 × 103 cells per well were seeded into 96-well plates and were incubated with Erastin or cell death inhibitors (Fer-1 or ZVAD) after being transfected for 24 h. Then, cells were added with 10 μL CCK-8 for 1 h. The absorbance was measured at 450 nm.
EdU proliferation assay
Cell proliferation was assessed by EdU Kit (KeyGen Biotech, Jiangsu, China) according to the manufacturer's instruction. Briefly, cells were treated by incubating with EdU reagent for 2 h at 37°C after being transfected for 48 h. Cells were rinsed with PBS twice and were fixed with 4% paraformaldehyde for 15 min. Then, cells were incubated with 0.5% Triton X-100 for 20 min at room temperature and were counterstained with Hoechst 33342 (diluted 1:2000) for 15 min. Images were captured by IX53 microscope. Subsequently, cells were counted manually from the EdU imaging data, and the proportion was calculated as follows: the count of cells in S phase/the total count of cells * 100%.
Cell cycle analysis
Cell Cycle Detection Kit (KeyGen Biotech) was used to detect the cell cycle progression according to the manufacturer's protocol. Briefly, after being treated as per the procedures above, cells were collected and washed with PBS thrice. Then, cells were fixed in precold 70% ethanol for fixation at 4°C overnight and then rinsed with PBS. One hundred microliters RNase A was added into cells at 37°C for 30 min. Cells were further stained with propidium iodide preventing from the light. The cell cycle was measured by a NovoCyte flow cytometer (ACEA Biosciences, San Diego, CA).
Dual-luciferase reporter system
Dual-luciferase reporter assay was performed by Dual-Luciferase Reporter Assay System (Promega, Madison, WI) following the manufacturer's instructions. Briefly, wild-type and mutant SLC7A11 promoters were synthesized and subcloned into the pGL3-Basic vector that was cotransfected with ETV4-OE and internal reference plasmid (phRL) into 293T cells. After being transfected for 48 h, cells were lysed and further incubated with luciferase substrate. The relative activity of the luciferase was assessed with the proportion of firefly luciferase to Renilla luciferase.
Lipid peroxidation
Lipid peroxidation was measured using a C11-BODIPY Kit (Thermo Scientific, Pittsburgh, PA) following the manufacturer's protocol. Briefly, the treated cells were incubated with 2 μM BODIPY 581/591 C11 for half an hour and then stained with DAPI. Images were obtained under IX53 microscope.
GSH assay
The treated cells were subjected to sonication, and the cell supernatant was collected for GSH analysis using a GSH Assay Kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions.
Mice xenograft
Six-week-old BALB/c female nude mice (HFK Bioscience, Beijing, China) were used to perform experiments in vivo. Mice were fed with chow ad libitum and housed in a temperature-controlled (24°C) under specific pathogen-free conditions. One hundred forty-four mice were randomly divided into four groups (36 mice in each group). TPC-1 cells were stably transfected with NC shRNA or ETV4-shRNA, and GLAG-66 cells were stably transfected with ETV4-OE or Vector. 5 × 106 cells were injected subcutaneously into the right armpit in mice. After 7 days, the diameter of tumors was measured every 3 days to calculate the tumor volume. Mice were sacrificed on the 22nd day after injection.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 8.0. One-way analysis of variance (ANOVA) or two-way ANOVA was used to compare the difference among multiple groups. Unpaired Student's t-test was used to compare the significance between the two groups except for the comparison of ETV4 expression between PTC clinical tumor tissues and adjacent normal tissues (paired Student's t-test). Data shown as proportions that were less than 30% or more than 70% were normalized by the square arcsine root transformation and then subjected to variance analysis. Results were shown as mean ± SD. The numbers of independent repeats for each experiment were as follows: in vitro, three; in vivo, six. p < 0.05 was considered as statistical significance.
Results
ETV4 expression was upregulated in PTC tissues and PTC cell lines
The mRNA expression levels of ETV4 in 30 pairs of PTC tissues, normal adjacent tissues, and PTC cell lines were determined by qRT-PCR. As shown in Figure 1A, ETV4 expression was higher in PTC tissues than in normal adjacent tissues. Compared with the normal thyroid cells (Nthy-ori 3-1), the relative expression of ETV4 was upregulated in PTC cells: IHH-4, TPC-1, and GLAG-66 cells (Fig. 1B). Moreover, TPC-1 cells exhibited the highest expression of ETV4, and GLAG-66 cells showed the lowest ETV4 expression. Thus, TPC-1 and GLAG-66 cells were selected for further ETV4 modification. The efficiency of the ETV4 interference was detected by qRT-PCR after 48 h (Fig. 1C). Two ETV4 siRNAs with the highest interference efficiency were chosen for the subsequent experiments. Meanwhile, we overexpressed ETV4 in GLAG-66 cells (Fig. 1D). The protein expression of ETV4 echoed the results of qRT-PCR (Fig. 1E; The uncut protein blots could be found in Supplementary Data).
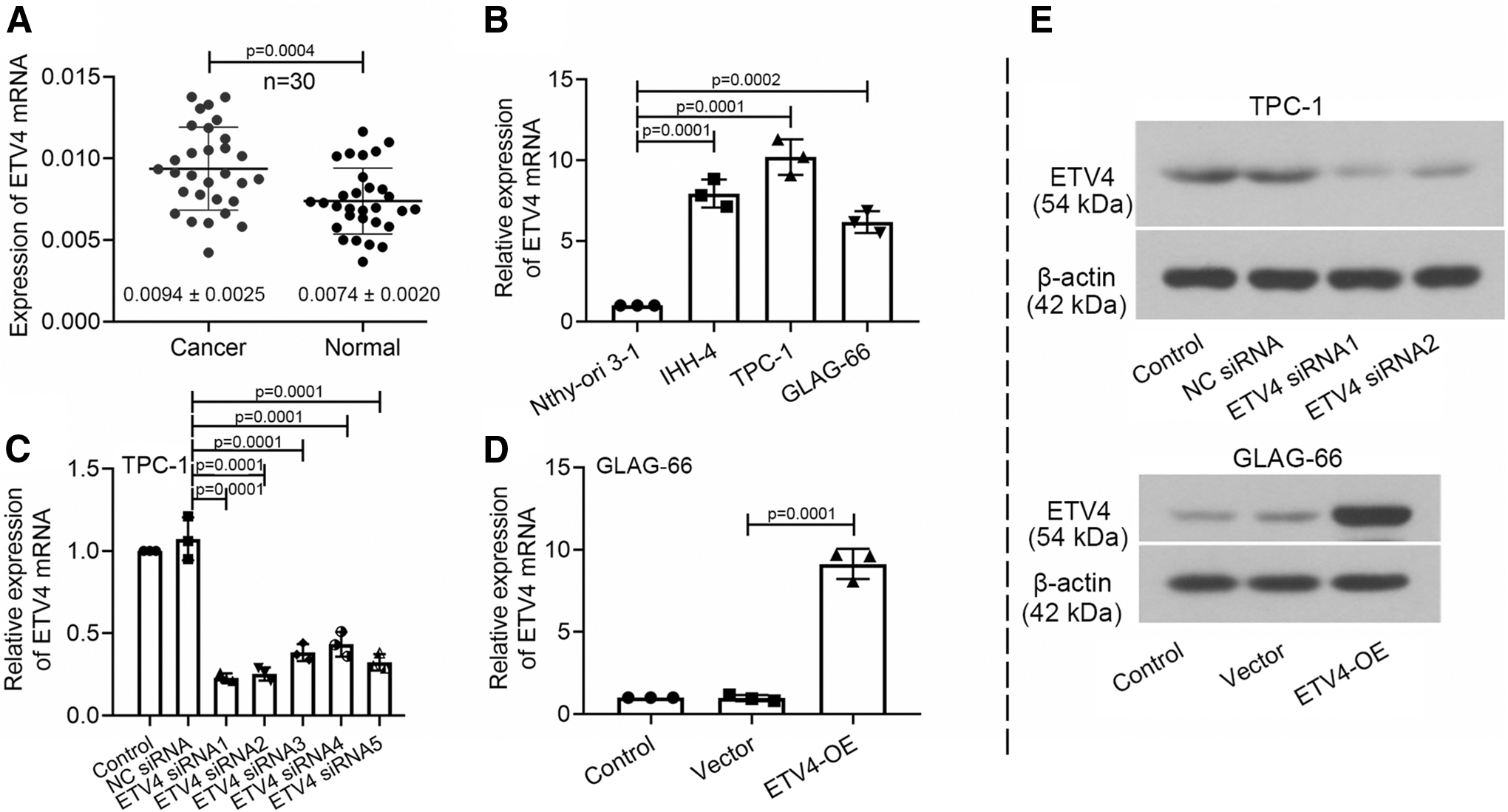
ETV4 expression was upregulated in PTC tissues and PTC cell lines.
The knockdown of ETV4 repressed cell proliferation and cell cycle progression of PTC cells
To determine whether ETV4 participated in cell proliferation in PTC progression, CCK-8 assay, colony formation assay, and EdU proliferation assay were conducted to PTC cells. Colony formation assay demonstrated that the ETV4 silence repressed the colony formation ability of TPC-1 cells, and ETV4 upregulation promoted the colony formation ability of GLAG-66 cells (Fig. 2A). Next, the growth curves of the CCK-8 assay showed that the cell proliferation rate was reduced after ETV4 siRNA transfection with TPC-1 cells, while the overexpression of ETV4 led to the opposite (Fig. 2B). Similarly, the EdU proliferation assay confirmed that cell proliferation was inhibited by ETV4 downregulation and was promoted by ETV4 overexpression (Fig. 2C). Next, flow cytometry was used to further explore the mechanism by which ETV4 contributed to cell proliferation. The results showed that the knockdown of ETV4 resulted in an elevation in the percentage of G1 phase cells accompanied by a reduction in S phase cells, and the overexpression of ETV4 obtained the contrary effects (Fig. 2D). Collectively, the knockdown of ETV4 repressed cell proliferation by arresting cells in G1 phase.
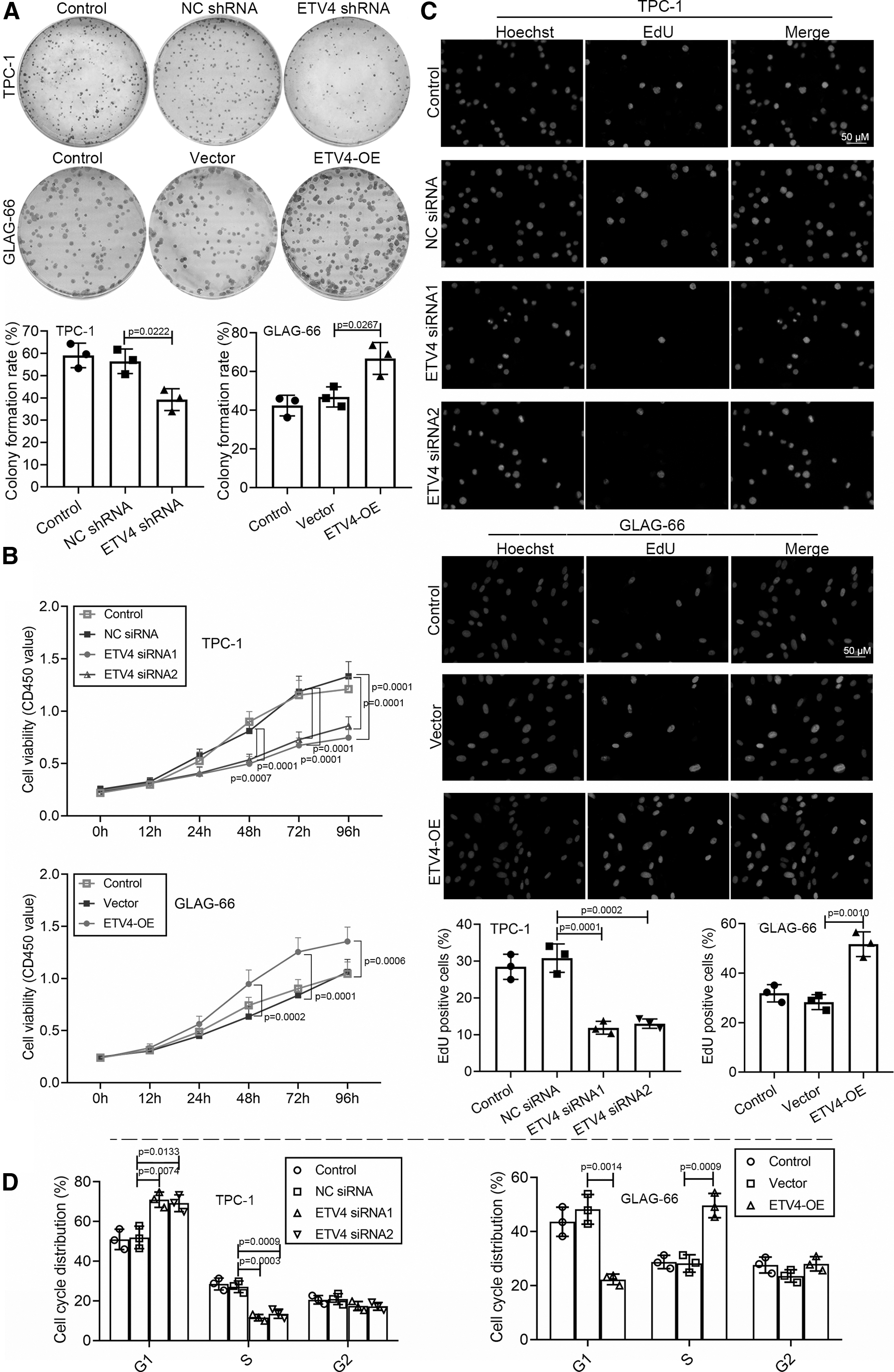
The knockdown of ETV4 repressed cell proliferation and cell cycle progression of PTC cells. For the ETV4 knockdown, TPC-1 cells were transfected with ETV4 siRNA1/2 or shRNA. For the ETV4 overexpression, GLAG-66 cells were transfected with the ETV4-OE vector.
ETV4 directly regulated the transcription of SLC7A11 in PTC cells
To provide a deep understanding of the underlying mechanism of ETV4 on the PTC progression, it is essential to explore the binding partner of ETV4. ETV4 was assessed and analyzed by the JASPER database, and two ETV4 binding sites “AGAGGAAGAC” (−2195 to −2186) and “TGAGGAAGCT” (−1995 to −1986) were predicted at the core regulation region of the SLC7A11 promoter (Fig. 3A). Subsequently, the results of the dual-luciferase reporter system confirmed that the mutation of the two binding sites reversed the increased luciferase activity (Fig. 3B). Then, we explored the regulatory mechanism of ETV4 on the SLC7A11 transcription by qRT-PCR and western blot. As revealed in Figure 3C, the SLC7A11 expression was decreased in TPC-1 cells induced by the interference of ETV4 and was increased in GLAG-66 cells with the overexpression of ETV4. And the protein expression of SLC7A11 echoed the results of qRT-PCR (Fig. 3D). These results suggested that ETV4 upregulated the SLC7A11 transcription by binding to the SLC7A11 promoter region directly.
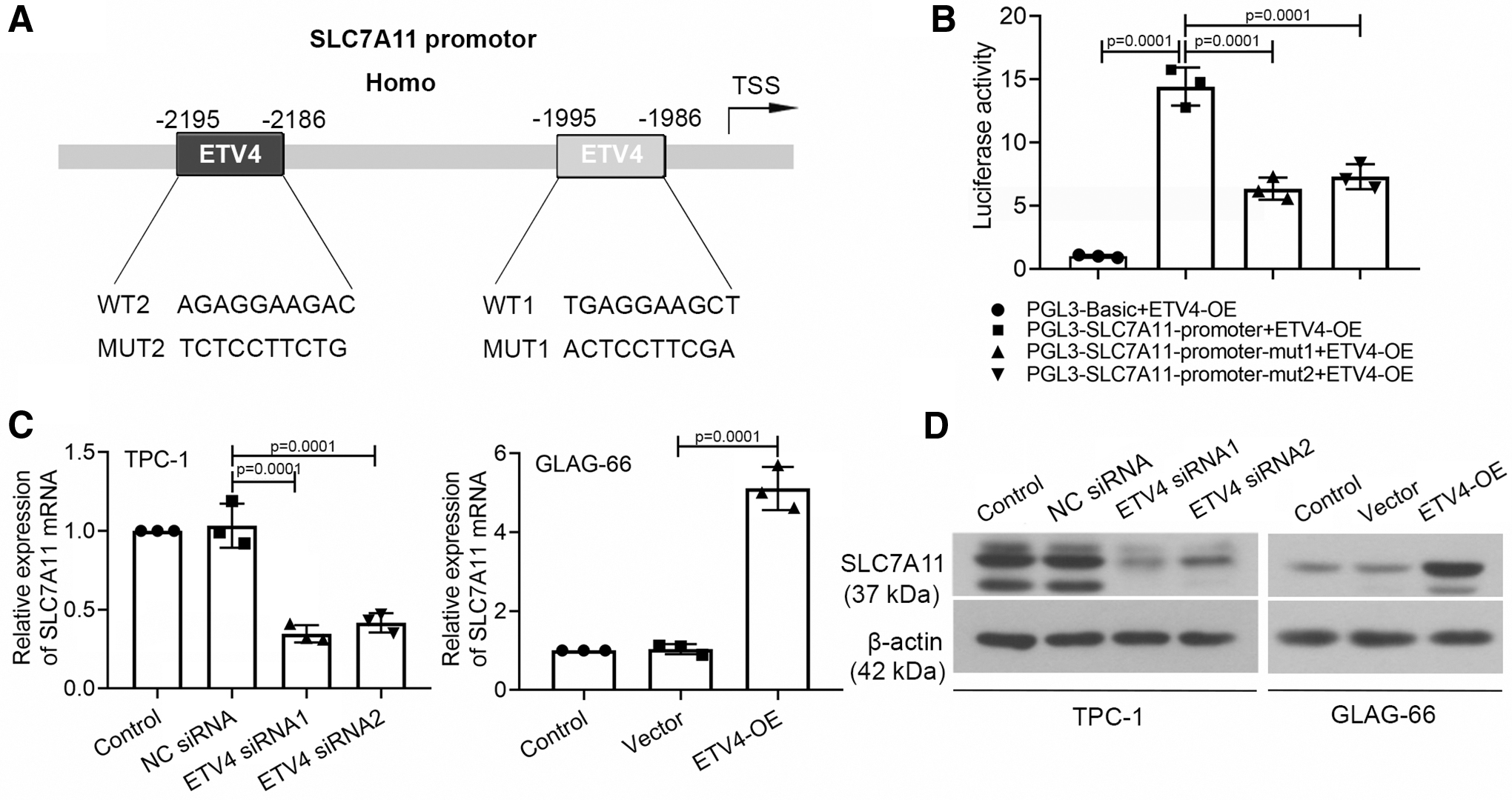
ETV4 directly regulated the transcription of SLC7A11 in PTC cells.
The knockdown of ETV4 inhibited the viability of PTC cells by promoting ferroptosis
SLC7A11 plays a vital role in cysteine uptake in the process of ferroptosis (Koppula et al., 2020). The essence of ferroptosis is the metabolic disorder of lipid oxides in cells, which disrupts the intracellular redox balance and then triggers cell death (Mou et al., 2019). To further explore the existence of ferroptosis and its role in PTC progression, we considered whether the ETV4 modification could rescue the ferroptosis progression. As shown in Figure 4A, the CCK-8 assay demonstrated that the ETV4 downregulation induced viability inhibition of TPC-1 cells was rescued in the presence of ferrostatin-1 or ZVAD-FMK in TPC-1 cells (Fig. 4A). The overexpression of ETV4 rescued the Erastin-induced viability repression and the increased density of green fluorescence (represented by C11-BODIPY staining), as well as the GSH level alleviation in GLAG-66 cells (Fig. 4B–D). The evidence indicated that viability inhibition of PTC cells induced by the knockdown of ETV4 was at least partly through the promotion of ferroptosis.
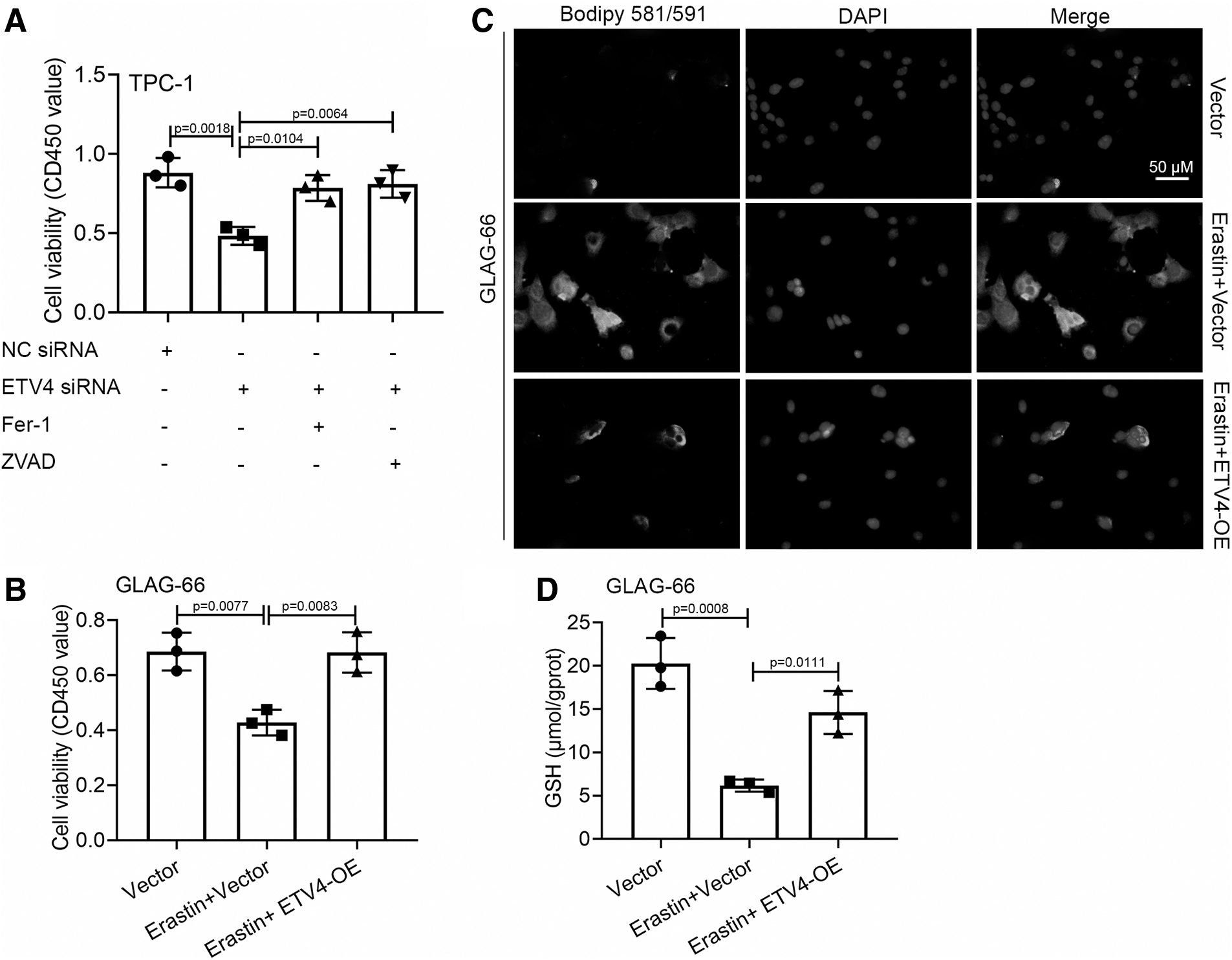
The knockdown of ETV4 inhibited the viability of PTC cells by promoting ferroptosis.
The knockdown of ETV4 inhibited the viability of PTC cells by facilitating ferroptosis upon SLC7A11 downregulation
We found that interfering in ETV4 expression inhibited the viability of PTC cells by promoting ferroptosis and downregulated the transcription of SLC7A11 as described above. Since SLC7A11 played a vital role in the cysteine uptake process of ferroptosis, we speculated that the ETV4-linked ferroptosis might be associated with the expression of SLC7A11. Thus, we cotransfected ETV4-OE with SLC7A11 siRNA into GLAG-66 cells. As revealed in Figure 5A and B, silencing SLC7A11 with specific siRNA reduced the protein expression of SLC7A11 and repressed the cell viability in ETV4-overexpressed GLAG-66 cells. The downregulated SLC7A11 weakened the cell viability and increased the lipid peroxidation level, as well as decreased the GSH level in Erastin-induced GLAG-66 cells with ETV4 overexpression (Fig. 5C–E). Our findings verified that the downregulated ETV4 inhibited the viability of PTC cells through facilitating ferroptosis upon the SLC7A11 downregulation.
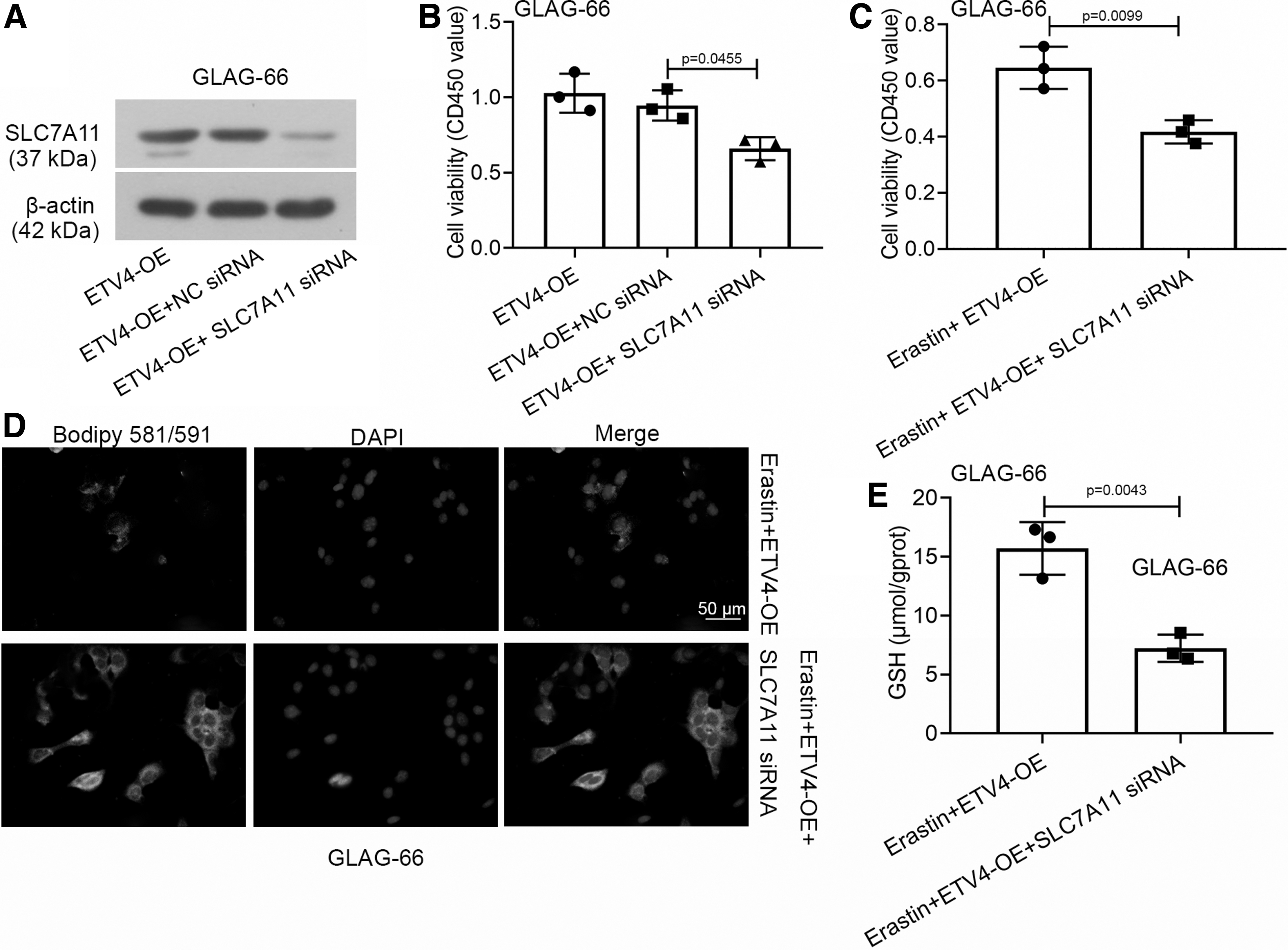
The knockdown of ETV4 inhibited the viability of PTC cells through facilitating ferroptosis upon SLC7A11 downregulation.
The knockdown of ETV4 suppressed the in vivo growth of PTC through the downregulation of SLC7A11
Based on the inhibition of downregulated ETV4 on PTC progression in vitro, we hypothesized that ETV4 may also affect the PTC progression in vivo. In support of the hypothesis, we injected mice with ETV4-OE vector stable transfected GLAG-66 cells or ETV4 shRNA stable transfected TPC-1 cells. As shown in Figure 6A–C, the growth of PTC xenografts in mice was promoted by the ETV4 overexpression and was suppressed by the knockdown of ETV4. Then, we verified the expression of ETV4 and SLC7A11 in the xenografts. Western blot demonstrated that ETV4 and SLC7A11 were low when expressed in tumor cells with the knockdown of ETV4 and were highly expressed in tumor cells with ETV4 overexpression (Fig. 6D). Together, our data showed that the knockdown of ETV4 suppressed the growth of PTC tumor through the downregulation of SLC7A11 in vivo.
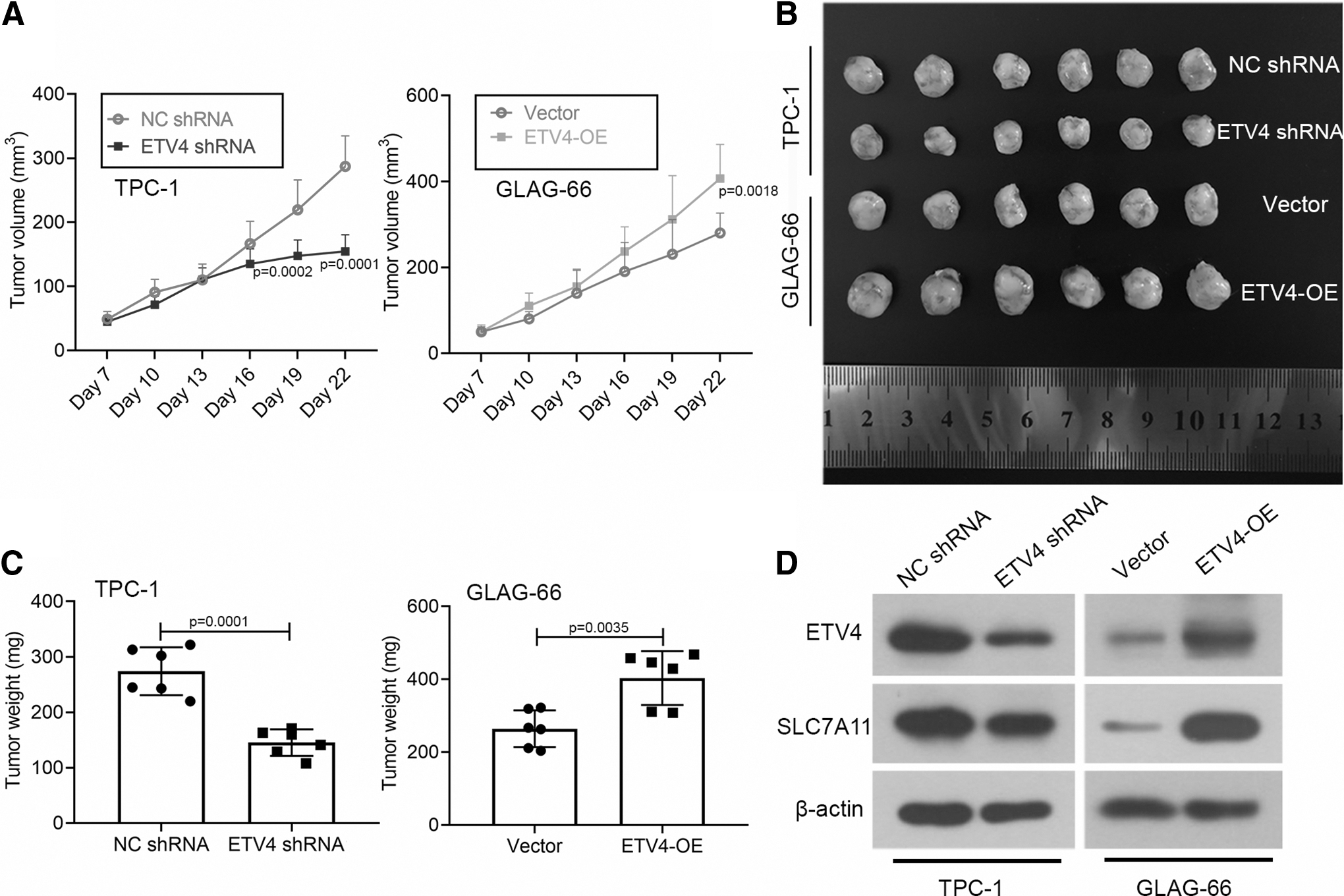
The knockdown of ETV4 suppressed the in vivo growth of PTC through the downregulation of SLC7A11. Mice were injected with ETV4-OE vector stable transfected GLAG-66 cells or ETV4 shRNA stable transfected TPC-1 cells.
Discussion
In the current study, we found that ETV4 was upregulated in PTC tissues and cells, and silencing ETV4 with specific siRNA inhibited cell proliferation by arresting PTC cells in the G1 phase in vitro. Further investigation demonstrated that ETV4 upregulated the SLC7A11 transcription by binding to the SLC7A11 promoter region directly. The knockdown of ETV4 inhibited the viability of PTC cells by promoting ferroptosis upon the downregulation of SLC7A11 in vitro and repressed the in vivo growth of PTC through the low expression of SLC7A11. Therefore, these results have significant implications on how ETV4 affected the PTC progression through ferroptosis.
ETV4 acts as an essential part of carcinogenesis by regulating the expression of the downstream relevant genes and is aberrantly expressed in various cancers, such as gastric, breast, lung, prostate cancer, as well as multiple myeloma (Dumortier et al., 2018; Cheng et al., 2019; Nicholas et al., 2019; Cai et al., 2020; Da Vià et al., 2020). ETV4 has been reported to have a high expression in PTC tissues and is linked with a poor prognosis (Song et al., 2019; Yu et al., 2020). In this study, we also confirmed that ETV4 expression was upregulated in PTC tissues and cell lines. Moreover, ETV4 regulates many essential cellular processes such as cell proliferation, metastasis, and apoptosis in carcinogenesis. Cheng et al. (2019) have proven that ETV4 enhances lung cancer cell proliferation and invasion by regulating the transcription of MSI2. Singsuksawat et al. (2018) have reported that the upregulated ETV4 accelerates the growth of xenograft tumor and facilitates the invasiveness of cholangiocarcinoma cells. In PTC, ETV4 is proven to enhance cell proliferation and metastasis by the upregulation of the MMP1 through binding to its upstream promoter (Yu et al., 2020). Herein, we artificially regulated the expression of ETV4 to further assess its effects on the PTC progression. The results showed that the knockdown of ETV4 resulted in the suppression of cell proliferation through the arresting cell cycle in the G1 phase in TPC-1 cells. And the overexpression of ETV4 facilitated cell proliferation by promoting G1/S transition in GLAG-66 cells.
To investigate the molecular mechanism of ETV4 in the PTC progression, it is critical to deeply assess the downstream signal of ETV4. Cosi et al. have reported that ETV4 is involved in the promoted cell proliferation through the downregulation of CDKN1A upon directly binding to a specific site of its promoter (Cosi et al., 2020). In PTC, the cancer invasiveness and progression might be mediated through BRAF-induced ETV4 upregulation by selectively binding to the mutant TERT promoter (Song et al., 2019). These studies suggest the possibility that the pro-tumor function of ETV4 might be mediated by the regulation of its target gene expression. Similarly, in this work, we found that two ETV4 binding sites were located at the core regulation region of the SLC7A11 promoter (located at −2195 to −2186 bp or −1995 to −1986 bp relative to the transcription start sites). Subsequently, the results of the dual-luciferase reporter system confirmed that the mutation of the two binding sites reversed the increased promoter activity, indicating that the regulation of ETV4 on SLC7A11 transcription in PTC cells was direct. Accumulating evidence has shown that SLC7A11 plays a significant role in lipid metabolism by facilitating cystine uptake for GSH synthesis, which is a key cofactor of the cellular ROS detoxification (Koppula et al., 2020). The high expression of SLC7A11 rescues cells from the ferroptotic cell death (Liu et al., 2019). Thus, we speculated that the effects of ETV4 on PTC progression might be associated with the ferroptosis mediated by SLC7A11.
Ferroptosis repressed the tumor growth and increased the sensitivity of chemotherapeutic drugs through the metabolic imbalance induced by GSH depletion in cancer cells (Lu et al., 2017). Different lines of evidence indicate that ferroptosis acts as a significant part in the inhibition of tumorigenesis. Jiang et al. (2015) have shown that p53 has a suppression of tumor growth by inhibiting the SLC7A11 expression and then induces ferroptosis, as well as apoptosis. Sui et al. (2018) have proven that the ferroptosis inducer ESL3 elicits colorectal cancer cell death through glutathione peroxidase 4 inactivation and ROS accumulation. Zhang et al. (2019) have demonstrated that the activity of tumor suppressor BAP1 is mediated by ferroptotic cell death through the downregulation of SLC7A11. Herein, our findings showed that the knockdown of ETV4 suppressed the viability of PTC cells through the ferroptosis facilitation by downregulating the SLC7A11 transcription in vitro and repressed the tumor development through the downregulation of SLC7A11 in vivo.
Ferroptosis is generally identified by cell death inhibitor rescue experiments. Ferroptotic cell death is inhibited by ferroptosis inhibitors and is not by inhibitors of other forms of cell death (Zhang et al., 2018). Interestingly, in this work, the ETV4 downregulation induced viability inhibition of TPC-1 cells was rescued in the presence of ferrostatin-1 or ZVAD-FMK in TPC-1 cells. And the ETV4 overexpression prevented Erastin-induced ferroptosis suppression by the decrease of lipid peroxidation and the accumulation of GSH level. Scholars have proven that ETV4 regulates cancer cellular processes, including proliferation, migration, and apoptosis. Chen et al. (2019) have reported that ETV4 could inhibit breast cancer cell apoptosis, which is regulated by the S100A8/A9-MCAM axis. The overexpression of ETV4 is testified to suppress cell apoptosis induced by sorafenib in hepatocellular carcinoma (Xiaohui et al., 2019). In this work, our findings suggested that ETV4 might be a negative regulator of various cell death processes, including apoptosis and ferroptosis.
Conclusion
ETV4 was highly expressed in PTC tissues and cells. The knockdown of ETV4 inhibited the PTC development by promoting ferroptosis upon SLC7A11 downregulation. Our findings prompted us to extend ferroptosis facilitation to PTC progression and suggested ETV4 as a potential target in PTC therapy.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was financially supported by grants from the Young Scholar Support Program 2018 of China Medical University (Grant number: QGZD2018061).
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.