
Abstract
Circular RNAs (circRNAs) are a class of noncoding RNAs closely related to the development and progression of various human cancers. However, it is unclear whether circRNAs play an important role in the development of bladder cancer. We utilized human circRNA array V2 microarrays to screen circRNA expression profiles in bladder cancer tissues. Bioinformatic tools including circBank, dbDEMC 2.0, miRCancer, TarBase v7.0, miRtarbase, TCGA–BLCA, Cytoscape-MCODE, String, ENCORI, and Venny 2.1 were then employed to construct the circRNA–miRNA–mRNA regulatory networks. In total, 105 upregulated circRNAs and 167 downregulated circRNAs (fold change >2 and p < 0.001) were filtered out. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of filtered dysregulated circRNAs disclosed that the circRNAs regulatory network was closely related with mRNA processing and cell cycle, etc. Further excavation analysis showed that seven differentially overexpressed circRNAs including hsa_circ_0000133, hsa_circ_0023610, hsa_circ_0005615, hsa_circ_0030162, hsa_circ_0077007, hsa_circ_0001140, and hsa_circ_0107031 were associated with bladder cancer invasiveness, and the cell cycle signal axis. has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 was finally clarified as a possible mechanism for bladder cancer progression. Based on results derived from multiple approaches, we identified that has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 signal axis may be involved in the invasion process of bladder cancer.
Introduction
Bladder cancer was the ninth most common type of cancer in the world (Antoni et al., 2017). According to the statistics of GLOBOCAN in 2020, there were ∼81,400 new cases worldwide and ∼13,050 deaths in the United States (Siegel et al., 2020). What is more surprising was that China accounted for 24% of newly diagnosed cases and 30% of the cancer-related deaths worldwide in 2020 (Cao et al., 2021). Cystoscopy and biopsy are still the good standard for the diagnosis of bladder cancer because ideal markers for bladder cancer are deficient. Surgical radical cystectomy combination chemotherapy is the main treatment on muscle-invasive and metastatic bladder cancer (Milowsky et al., 2016).
The survival rate of patients with bladder cancer depends mainly on the stage of cancer at the time of diagnosis (Westergren et al., 2019). Although the mortality rate for patients diagnosed with nonmuscle invasive bladder cancer (NMIBC) is far from satisfactory, patients with muscle invasive bladder cancer (MIBC) have a poorer prognosis and are more likely to progress to metastasis (Kamat et al., 2016; Dy et al., 2017). Therefore, effective diagnosis of biomarkers will play a very positive role in improving the prognosis and response of patients.
Circular RNAs (circRNAs) are broadly expressed in a variety of organisms. CircRNAs are closed long non-coding RNAs characterized by linking the 3′ and 5′ termini form a covalently closed loop structure, which play an imperative role in gene expression or translation of regulatory proteins. (Yu and Kuo, 2019). Unfortunately, circRNAs attracted little attention for long time as they have always been regarded as accidental misplacing by-products or spliced intermediates of post-transcription (Sanger et al., 1976). With the improvement of the new-generation sequencing technology, the accurate structure and potential function of circRNAs have been recognized and have been shown to be closely related to the development and progression of human tumors and other diseases (Cooper et al., 2018).
Tumors such as gastric, colorectal cancer, and hepatocellular carcinoma showed significant differences in the expression profiles of circRNAs (Taborda et al., 2017; Vidal et al., 2017; Xu et al., 2017). Recent studies have shown that circRNAs were involved in various stages of the carcinogenesis process (Chen and Huang, 2018), suggesting an important regulatory role in cancer pathogenesis and metastasis. It is widely used for early stage risk stratification and survival prediction due to its rich expression, conservation, stability, and tissue specificity.
The aim of this research was to explore the expression profiles of circRNAs in bladder cancer, to construct a global circRNA–miRNA–mRNA regulatory network, to investigate the differential circRNAs between NMIBC and MIBC specimens, and to elucidate their potential regulatory functions, finally providing new ideas or targets for effectively monitoring and treating bladder cancer patients.
Materials and Methods
Clinical bladder cancer specimen collection
This research was approved by the ethics committee for biomedical research of 900 Hospital of the Joint Logistics Team (The institutional approval number for this study is 2021-006). Human bladder cancer specimens that are 10 bladder cancer specimens (5 patients with NMIBC and 5 patients with MIBC) and 5 patients with benign prostatic hyperplasia or bladder stones but no history of bladder cancer (control group) were collected from the department of urology in 900 Hospital (see Supplementary Table S1). All the specimens were quickly cryopreserved in nitrogen canister in vitro for subsequent experiments. All patients were informed and gave written informed consents for research purpose.
RNA isolation
Total RNA originated from cryopreserved tissues in nitrogen canister was isolated by TRIZol agent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The concentrations of the RNA samples were then determined by the NanoDrop ND-1000 (Nano Drop Thermo, Wilmington, DE) instrument. The integrity of RNA was also assessed by electrophoresis on a denaturing agarose gel.
Labeling and hybridization
The expression levels of circRNAs in 15 bladder cancer specimens were detected by Arraystar Human circRNA Array V2 (8x15K; Arraystar, Shanghai, China). Total RNA from each sample was quantified using the NanoDrop ND-1000. Sample preparation and microarray hybridization were performed based on the Arraystar's standard protocols. In brief, total RNAs were digested with Rnase R (Epicentre, Inc.) to remove linear RNAs and enrich circRNAs. Then, the enriched circRNAs were amplified and transcribed into fluorescent cRNAs utilizing a random priming method (Arraystar Super RNA Labeling Kit; Arraystar). The labeled cRNAs were hybridized onto the Arraystar Human circRNA Array V2 (8x15K; Arraystar). After having washed the slides, the arrays were scanned by the Agilent Scanner G2505C.
Microarray data analysis process
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the R software Limma package. After quantile normalization of the raw data, low intensity filtering was performed. Differentially expressed circRNAs with statistical significance between two groups were identified through Volcano Plot filtering. Differentially expressed circRNAs between two samples were identified through fold change (FC) filtering. Hierarchical clustering was performed to show the distinguishable circRNAs expression pattern among samples.
Construction of circRNA–miRNA regulatory network
To establish the differential circRNA–miRNA regulatory network diagram of bladder cancer, the circRNA–microRNA interaction was predicted using Arraystar's home-made miRNA target prediction software, and circRNA–miRNA regulatory network diagram was plotted using Cytoscape 3.7.2.
KEGG pathway and GO terms analysis
The possible biological pathways of all differentially expressed circRNAs were explored by circRNAs–miRNAs–mRNAs regulatory axis. First, the conserved miRNA binding site (circRNAs–miRNAs) corresponding to differentially expressed circRNAs was obtained using Arraystar's home-made miRNA target prediction software, and then the experimentally verified interaction of miRNAs–mRNAs was obtained by using DIANA-TarBase V7.0 (
Screening of differentially overexpressed circRNAs in MIBC compared with NMIBC
When comparing two groups of profile differences (such as MIBC vs. NMIBC, MIBC vs. normal, or NMIBC vs. normal), the FC (i.e., the ratio of the group averages) between the groups for each circRNA was computed. See more specific information of patients on Supplementary Table S1. The statistical significance of the difference may be conveniently estimated using t-test. circRNAs having FCs ≥2 and p-values <0.05 were selected as the significantly differentially expressed. Differentially overexpressed circRNAs among the three groups were analyzed using Venny 2.1 (
TCGA–BLCA data preprocessing
RNA-seq and miRNA-seq of the TCGA–BLCA transcriptome were downloaded by using GDC Data Transfer Tool from The Cancer Genome Atlas (TCGA). Standardization and normalization of raw data and subsequent differentially expressed analysis processing were performed using the R software edgeR package.
Prediction for circRNA–miRNA–mRNA pathways
To establish differential circRNA–miRNA regulatory network diagram of differentially overexpressed circRNA–miRNA–mRNA in MIBC compared with NMIBC, the first step in this process was to utilize circBank (
The sets of circBank–miRNAs, dbDEMC 2.0-miRCancer–miRNAs, and TCGA–miRNAs were intersected to acquire downstream target miRNAs of differentially overexpressed circRNAs in MIBC compared with NMIBC (circRNAs–miRNAs). The circRNAs–miRNAs target mRNAs that were experimentally verified were identified using DIANA-TarBase V7.0 and miRtarbase (TarBase-miRtarbase–mRNAs). At the same time, the differential expressed upregulated mRNAs of TCGA–BLCA were screened (TCGA–mRNAs) (p < 0.05, log2FC >2). At last, the sets of TarBase-miRtarbase–mRNAs and TCGA–mRNAs were intersected to acquire circRNA–miRNA–mRNA pathways (circRNAs–miRNAs–mRNAs).
Construction of circRNAs–miRNAs–hubgene network
According to the target mRNAs list of circRNAs–miRNAs, the functional protein association network was plotted using STRING (
Delineation of circRNA–miRNA–mRNA core regulatory network related to MIBC
According to the circRNAs–miRNAs–hubgene network and combining with TCGA, DIANA-TarBase v7.0, and miRtarbase, core regulatory network of circRNA–miRNA–mRNA signal axis was plotted using Cytoscape 3.7.2.
Quantitative real-time polymerase chain reaction
The expression of circRNA was detected by quantitative real-time polymerase chain reaction (qRT-PCR) by applying the SYBR® Green Master Mix (Thermo Fisher Scientific, Waltham, MA). Actin was used as internal reference control. The specific primer sequences for human circRNA are as follows. hsa_circTPT1_003: 5′-TGACTCGCTCATTGGTGGAA-3′ (forward); 5′-CAGCCCCTGTCATAAAAGGT-3′ (reverse). Actin: 5′-CATGTACGTTGCTATCCAGGC-3′ (forward); 5′-CTCCTTAATGTCACGCACGAT-3′ (reverse). All experiments were conducted according to the manufactures' instructions and data are expressed as bar in the mean ± SD in triplicate.
Analysis of the correlation between the expression of miRNAs and mRNA in bladder cancer
ENCORI (
Statistical analysis
Statistical analysis was conducted by using R software (version 3.4.4) and the GraphPad Prism (version 7.0) software. Student's t-test was employed to compare the differences between groups. p-Values <0.05 was considered statistically significant.
Results
Identification of differentially expressed circRNA profiles
Quantile normalization of the raw data was used to erase individual differences between samples (Fig. 1A). Hierarchical clustering is one of the simplest and widely used clustering techniques for analysis of gene expression data. Cluster analysis arranges samples into groups based on their expression levels, which allows us to hypothesize about the relationships among samples. The results of hierarchical cluster analysis of differentially expressed circRNAs among 15 samples displayed that there was a significantly differentially expressed circRNA profile between the bladder cancer group and the normal control group, and mainly the downregulated circRNAs (Fig. 1B).
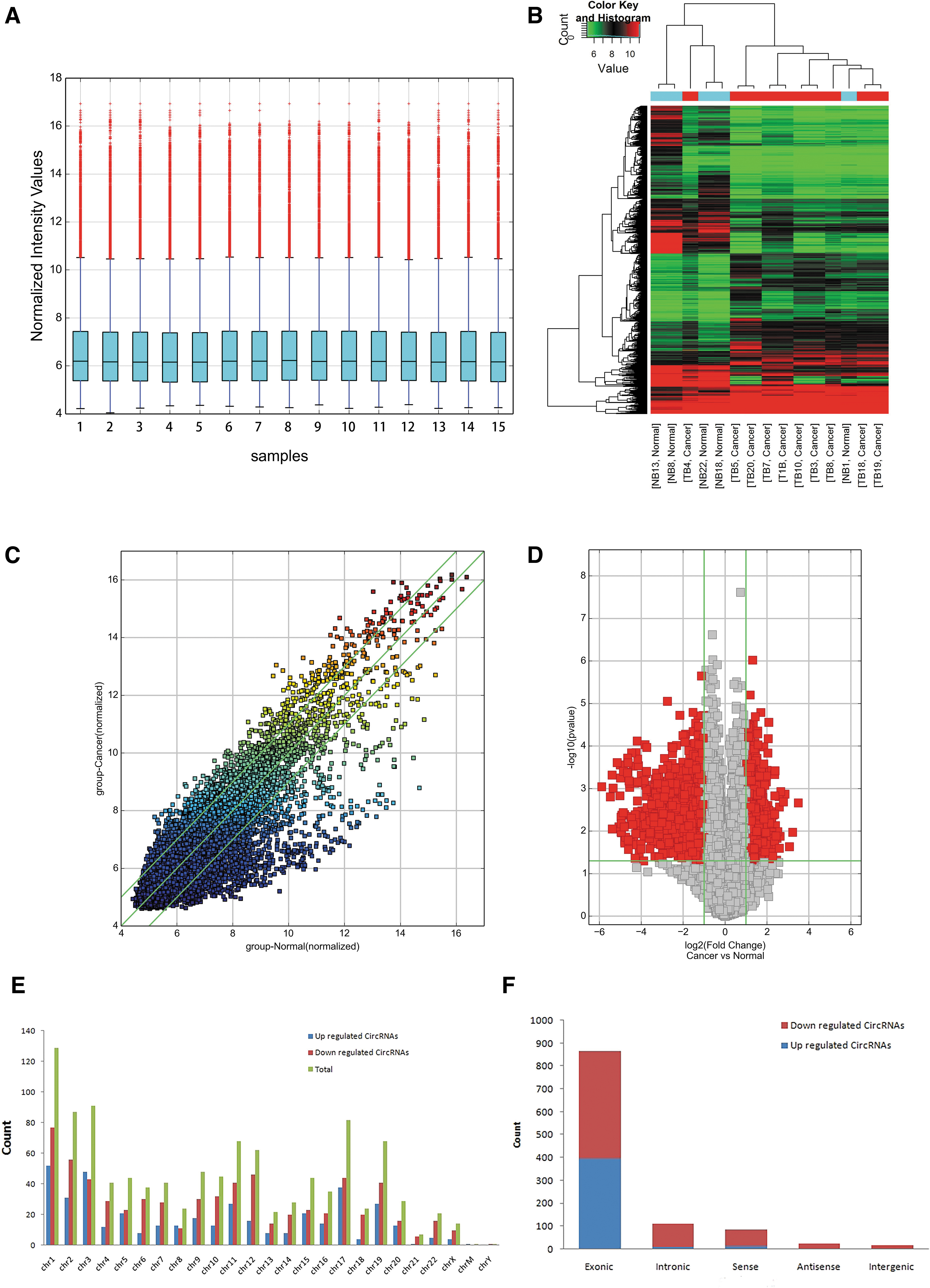
Differences and distribution characteristics of circRNA expression profiles between bladder cancer tissues and counterpart normal tissues.
A total of 13,048 target circRNAs were detected in 15 specimens using microarray probes. Differentially expressed circRNAs between specimens were shown through FC filtering (Fig. 1C). And differentially changed circRNAs with statistical significance between two groups were displayed through Volcano Plot filtering (Fig. 1D). The results showed that 272 differentially expressed circRNAs were screened comparing with adjacent noncarcinoma tissues. (|FC| > 2, p < 0.001).
Among which 105 circRNAs were upregulated while 167 circRNAs were downregulated. And the down-expression group is more common than the high-expression group (see Supplementary Table S2). The differentially expressed circRNAs in bladder cancer are distributed in almost all human chromosomes, and they, respectively, originate from the “Exonic,” “Intronic,” “Sense overlapping,” “Antisense,” and “Intergenic” (Fig. 1E, F). As a result, the differentially expressed circRNAs are mostly derived from exons that encode proteins.
Construction of the circRNA–miRNA network and prediction of biological function in bladder carcinoma
Recent pieces of evidence have demonstrated that circRNAs can recognize and adsorb miRNAs, through the principle of complementary base pairing to play the role of miRNA sponge, and then turn the level of miRNA-mediated regulation of gene expression. Therefore, we utilized the software for miRNA target prediction made in Arraystar to predict differentially expressed circRNA target miRNA through conserved seed-matching sequence in bladder cancer (see Supplementary Table S3), and plotted the following with Cytoscape (Fig. 2A). The results showed that differentially expressed circRNA may be major driving forces of the development and progression of bladder cancer through the function by sponging miRNAs.
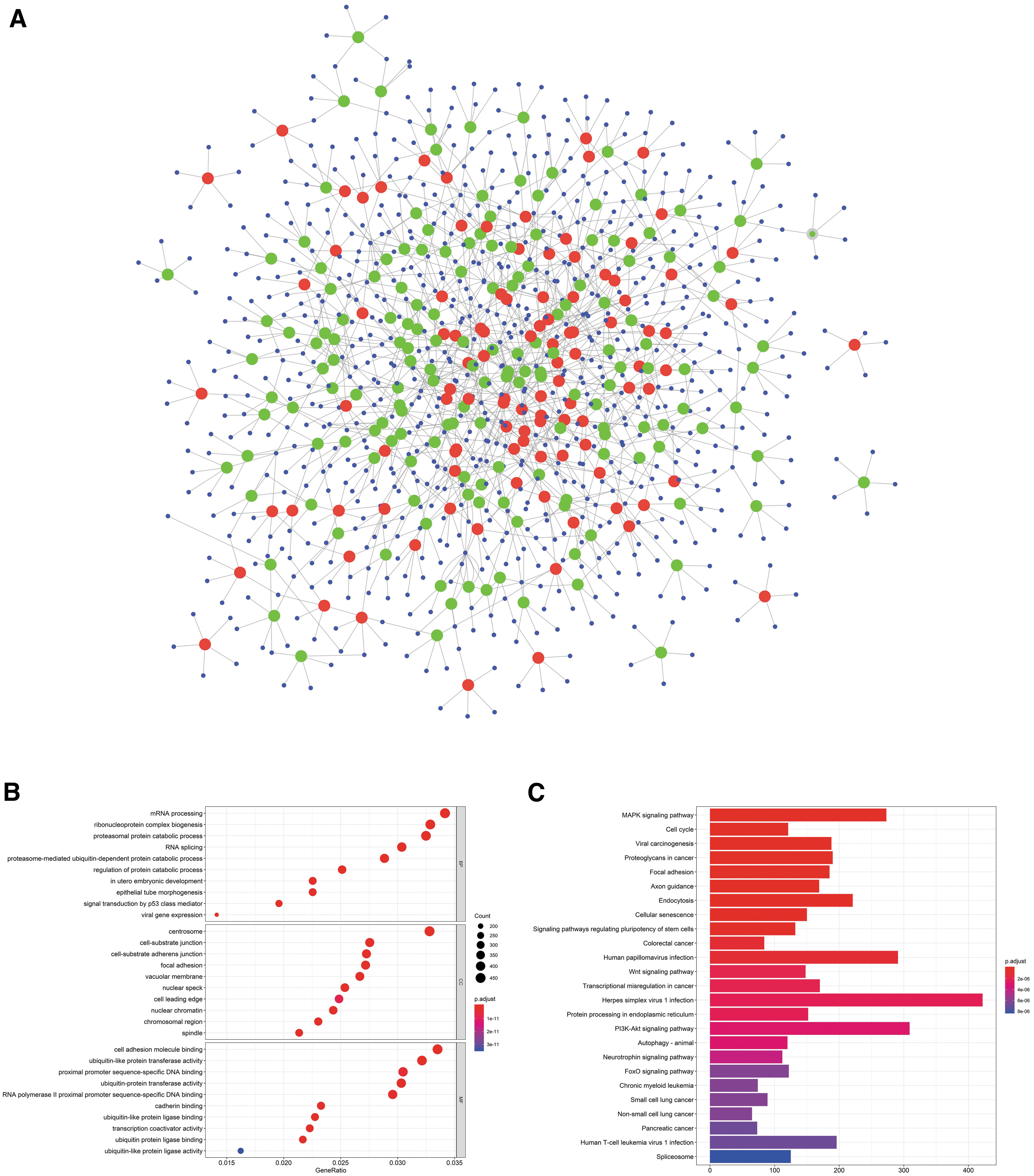
Construction of the circRNA–miRNA network and analysis of GO and KEGG.
To predict the carcinoma-associated pathways of circRNA–miRNA–mRNA regulated network, both GO and KEGG pathway analyses were performed by using clusterProfiler R package (Fig. 2B, C). GO analysis displayed that differentially expressed circRNAs might be involved in 1181 biological processes (BPs), 217 cellular components (CCs), and 128 molecular functions (MFs). It mainly involves mRNA processing, RNA splicing, proteasome, nucleosome, ubiquitin transferase activity, etc. The result of the KEGG pathway analysis showed that these circRNAs were also implicated in MAPK, cell cycle regulation, proteoglycans in cancer, etc.
Determining the differentially overexpressed circRNAs in MIBC compared with NMIBC
To identify circRNAs related to MIBC, we focused on the analysis of differentially overexpressed circRNAs between MIBC and NMIBC, MIBC and normal, and between NMIBC and normal in microarray. Differentially expressed circRNAs between specimens were shown through FC filtering (Fig. 3A). And differentially changed circRNAs with statistical significance between two groups are displayed through Volcano Plot filtering (Fig. 3B). As results, 526 differentially overexpressed circRNAs were detected between MIBC group and NMIBC group, 977 differentially overexpressed circRNAs were detected between MIBC group and normal group, and 1361 differentially overexpressed circRNAs were detected between NMIBC group and normal group (FC >2, p < 0.05).
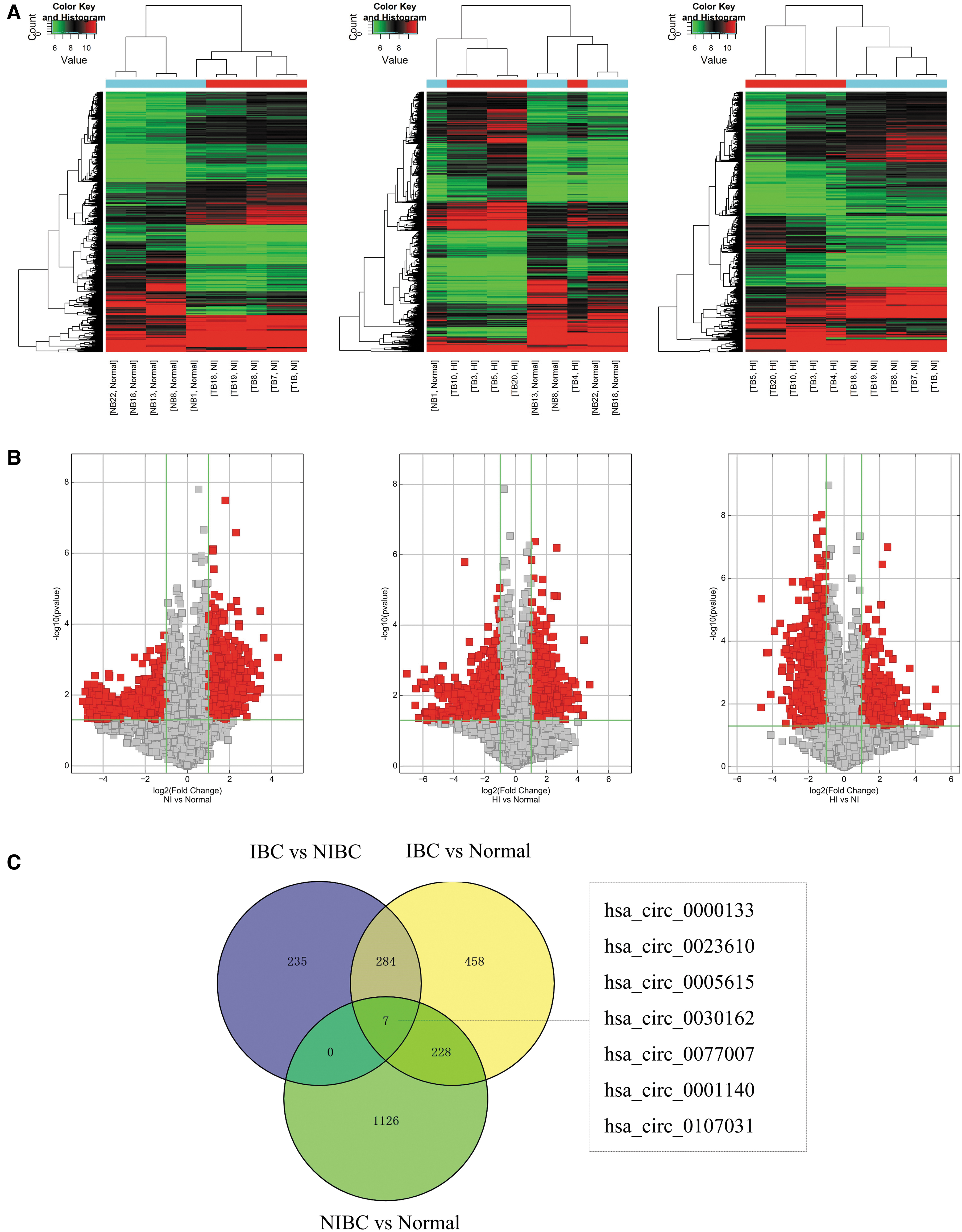
Screening associated with invasive bladder cancer of differentially overexpressed circRNAs.
After obtaining the list of three sets of differentially overexpressed circRNAs, the seven co-overexpressed circRNAs were found from Venn diagram of intersections of three sets. As shown in Figure 3C, they included the following seven circRNAs: hsa_circ_0000133, hsa_circ_0023610, hsa_circ_0005615, hsa_circ_0030162, hsa_circ_0077007, hsa_circ_0001140, and hsa_circ_0107031. The differential expression of them was performed. As shown in Supplementary Figure S1, seven circRNAs were significantly different between two groups (MIBC vs. NMIBC, MIBC vs. normal, and NMIBC vs. normal).
Construction of differentially overexpressed circRNA–miRNA–mRNA associations in MIBC compared with NMIBC
CircRNA could act as a sponge to adsorb peripheral miRNAs, to sequestering the miRNAs target downstream mRNA, and the play a fatal role in transcription regulation. So, the seven differentially overexpressed circRNAs related to invasive bladder cancer have similar function? To facilitate the following study, the circRNA–microRNA interaction was predicted with circBank based on TargetScan and miRanda. The seven differentially overexpressed circRNAs related to invasive bladder were analyzed in detail. Among them, the sequences of hsa_circ_0005615, hsa_circ_0023610, hsa_circ_0001140, hsa_circ_0030162, and hsa_circ_0000133 may possess more miRNA binding locus, therefore, they are more likely to play a crucial role in the regulation of competitive endogenous RNA (ceRNA) network.
Subsequently, to get a more comprehensive and broader view of circRNA target miRNAs, three sets circBank–miRNAs, dbDEMC 2.0-miRCancer–miRNAs, and TCGA–miRNAs were intersected in this section. All predicted target miRNAs are listed in Supplementary Table S4. As shown in Figure 4A, circRNAs related to invasive bladder cancer may target 10 miRNAs that are hsa-miR-145-5p, hsa-miR-5698, hsa-miR-495-3p, hsa-let-7c-3p, hsa-miR-3199, hsa-miR-504-5p, hsa-miR-654-5p, hsa-miR-204-5p, hsa-miR-218-5p, and hsa-miR-338-5p.

Construction of circRNA target miRNAs and miRNA target mRNAs and identification of core cluster of PPI.
So how to predict the miRNA target genes? The set of TarBase-miRtarbase–mRNA analysis showed that miRNA may target 3967 genes, which have been validated by experiments. And other 2991 genes were from TCGA–mRNA (p < 0.05, log2FC >2). In total, 361 genes were gained when TarBase-miRtarbase–mRNAs and TCGA–mRNAs were intersected by using Venny 2.1 (Fig. 4B) (Supplementary Table S5). To construct a protein–protein interaction (PPI) network for 361 genes, the STRING database was utilized, and then the PPI was explored deeply by using Cytoscape-MCODE plugin to identify the crucial modules in the circRNA–miRNA–mRNA associations.
The cluster with a score of 37.022 was recognized, which included KIF18A, CENPF, TRIP13, CCNE2, KPNA2, ASPM, E2F8, BIRC5, WDHD1, CDC6, CCNF, CENPM, ATAD2, CCNA1, CDK1, CDCA2, CEP55, CDK2, CCNA2, KIF22, TOP2A, CDCA8, SGOL1, INCENP, CDT1, RACGAP1, MCM3, MKI67, FBXO5, CCNB1, CKS2, RAD51, NCAPD2, OIP5, BRCA1, SMC4, POLQ, HJURP, FOXM1, MCM2, TICRR, KIF15, ZWILCH, KNTC1, HELLS, and CDC25A (Fig. 4C). This will lay a substantial foundation for the establishment of core circRNA–miRNA–mRNA associations related to invasive bladder cancer progression.
Prediction of biological function of circRNA–miRNA–mRNA network
To explore possible BP of 46 genes in core cluster, both GO and KEGG pathway analyses were performed by using clusterProfiler R package (Fig. 5A, B). GO analysis displayed that differentially expressed circRNAs might be involved in 265 BPs, such as chromosome segregation, DNA replication, and cell cycle; 47 CCs, such as chromosomal region and centromeric region; and 12 MFs, such as cyclin-dependent protein kinase activity and chromatin binding. The result of the KEGG pathway analysis showed that these circRNAs were also implicated in 21 signaling pathways, such as cell cycle, cell senescence, DNA replication, and p53 signaling pathway.

The analysis of GO and KEGG pathway for 46 genes in core cluster.
Construction of circRNA–miRNA–mRNA signaling axis
The diagram of circRNA–miRNA–mRNA signaling axis was plotted by using Cytoscape 3.7.2. As show in Figure 6, the network consists of 4 circRNAs (hsa_circ_0000133, hsa_circ_0023610, hsa_circ_0005615, and hsa_circ_0030162), 10 miRNAs (hsa-miR-204-5p, hsa-miR-654-5p, hsa-miR-504-5p, hsa-miR-5698, hsa-miR-338-5p, hsa-miR-218-5p, hsa-miR-145-5p, hsa-miR-495-3p, hsa-let-7c-3p, and hsa-miR-3199), and several mRNAs. The tight correlation and mutual regulation were well illustrated in this network.
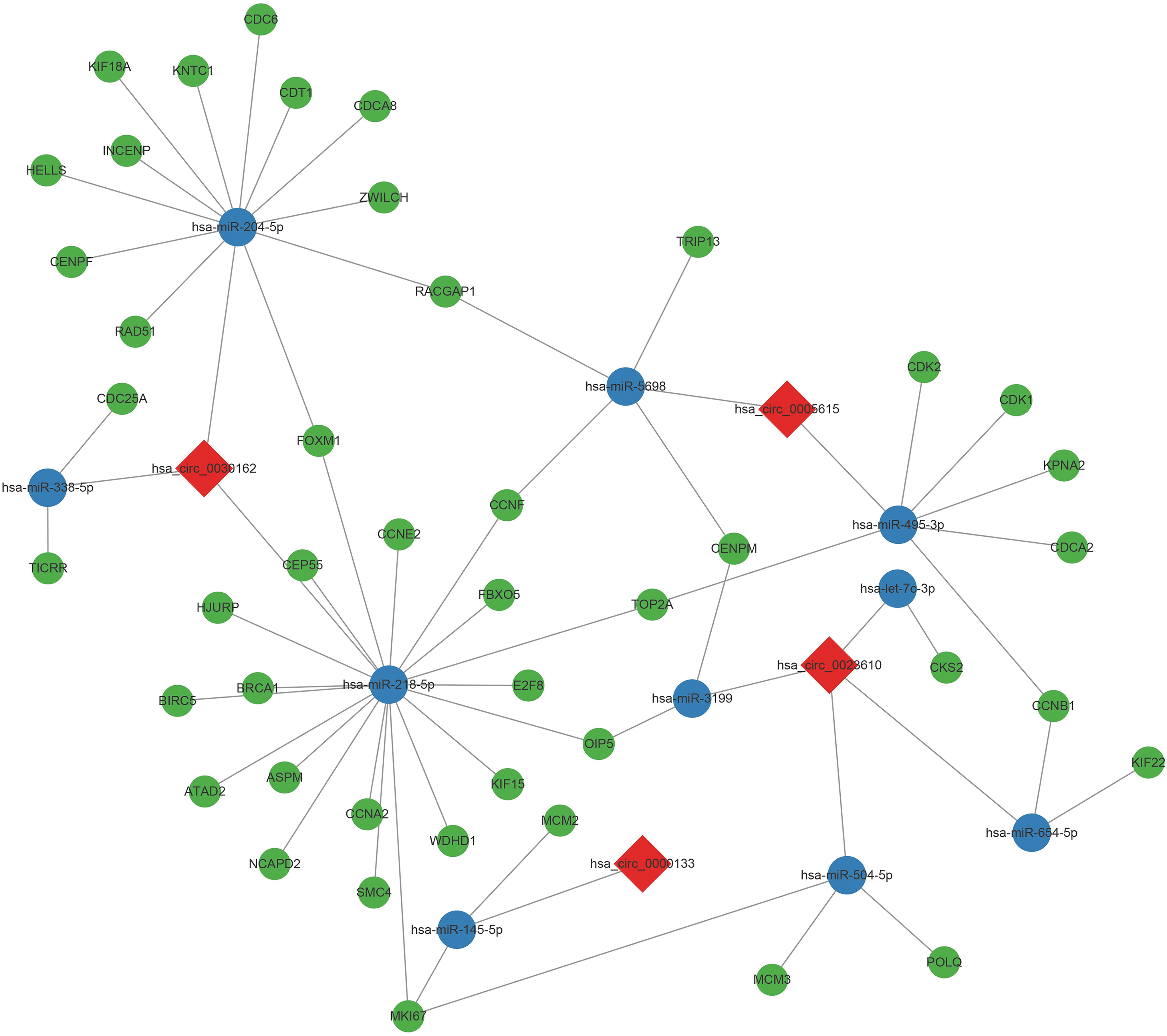
The regulatory network of circRNA–miRNA–mRNA signaling axis. Red, circRNA; blue, miRNA; green, mRNA. Color images are available online.
The initial establishment of has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 signaling axis in bladder cancer
To further identify key circRNA, the circRNA–miRNA interaction was predicted with Arraystar's home-made miRNA target prediction software through conserved binding miRNA sequence of circRNA. With alignment analysis of four circRNAs, only hsa_circ_0030162 (namely hsa_circTPT1_003) did meet the screening conditions. Schematic illustration of hsa_circTPT1_003 was inquired by using CSCD (

The initial establishment of has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 signaling axis.
It was predicted that hsa_circTPT1_003 could harbor two conserved sites (75–94, 252–272) of hsa-miR-218-5p by miRNAs seed sequence matching (Fig. 7C). Then, preliminary study of the expression of hsa_circTPT1_003 and correlation between that and target genes in TCGA–BLCA data was conducted. The results showed that significant trend was discovered for hsa_circTPT1_003 targeting hsa-miR-218-5p in bladder cancer and hsa-miR-218-5p could negatively regulate CCNE2 or SMC4 (Fig. 7D). In addition, we also determined the expression of SMC4 and CCNE2 in TCGA–BLCA paired samples. The result further supported the association between circTPT1_003 and hsa-miR-218-5p (Supplementary Fig. S2).
Discussion
In recent years, circRNAs have received more attention in regulating gene expression functions (Esteller, 2011; Verduci et al., 2019). Salzman et al. identified thousands of circRNAs derived from normal and malignant human cells by RNA sequencing technology (Salzman et al., 2012). Different from linear RNA, circRNAs form a covalently closed continuous loop to prevent degradation by RNA exonuclease. This contributes to stability and abundance of circRNAs in cells (Jeck et al., 2013).
The extracellular circRNAs expression can be regulated by exosome removal (Lasda and Parker, 2016). Recent studies have shown that circRNAs seem to act as an miRNA sponge, which is partially attributable to the formation of the ceRNA network (Hansen et al., 2013). The tissue specificity of circRNA expression under pathological conditions indicates that they may play an important role in human cancer (including bladder cancer) and other diseases (Salzman et al., 2013; Cong et al., 2019).
In this study, a total of 861 significantly differential expression circRNAs were detected in bladder cancer by using Arraystar human circRNAs microarray compared with the adjacent noncarcinoma tissues, including 364 upregulated circRNAs and 497 downregulated circRNAs. Interestingly, more circRNAs were significantly downregulated in bladder cancer. Zhong et al. (2016) also screened the differential circRNAs expression profiles in bladder cancer through the same technology and 285 upregulated circRNAs and 184 downregulated circRNAs were identified, however, the results were a trend toward overexpression of circRNAs in bladder cancer.
The reason for the mentioned might be due to the difference in type of clinical specimens. Sun et al. (2019b) elucidated that overexpression of circCEP128 manipulated BPs such as migration, proliferation, and apoptosis in bladder cancer cells through MAPK signaling pathway. In our study, the critical biological functions of circRNA–miRNA–mRNA axis in crucial signal pathways were assessed by GO and KEGG pathway analyses. The results showed that the differential expression of circRNAs was mainly related to MAPK, cell cycle signal pathways (Fig. 2B).
In most cases, the treatment methods for bladder cancer were mainly based on the staging and grading at the first diagnosis. So, it was particularly important to identify the differential expression and biological functions of circRNAs in different stages of the progression of bladder cancer. Seven differentially overexpressed circRNAs were reaped in MIBC compared with NMIBC, including hsa_circ_0000133, hsa_circ_0023610, hsa_circ_0005615, hsa_circ_0030162, hsa_circ_0077007, hsa_circ_0001140, and hsa_circ_0107031.
Bioinformatic tools including circBank, dbDEMC 2.0, miRCancer, TarBase v7.0, miRtarbase, TCGA–BLCA, Cytoscape-MCODE, String, ENCORI, and Venny 2.1 were utilized for constructing circRNA–miRNA–mRNA regulatory networks (Fig. 6). The hsa-mir-145-5p has been widely reported in bladder cancer among the 10 miRNAs regulated by differentially expressed circRNAs. circCEP128 and circ_0058063 promote bladder cancer progression by sponging miR-145-5p (Sun et al., 2019a, 2019b). Fujii et al. (2015) found that the expression of miR-145-5p was significantly downregulated in high-grade urothelial carcinoma, and overexpressed miR-145-5p suppressed cell proliferation of bladder cancer.
These suggested that the seven differentially overexpressed circRNAs may act as an invasion-promoting gene in bladder cancer. However, further in-depth research was required for other nine miRNAs in bladder cancer. Interestingly, BPs of 46 genes in core cluster were tightly associated with cell cycle (Fig. 5). Similarly, Dandan's research showed that circRNAs are involved in cell proliferation of bladder cancer (Wang et al., 2018). It is indicated that the differentially overexpressed circRNAs in MIBC compared with NMIBC may participate in cell cycle of bladder cancer by sponging miRNAs.
hsa_circTPT1_003, located in chr13:45911303–45914319, was derived from host TPT1 gene. TPT1 was classified as an oncogene that encoded protein involved in various biological functions, including cell growth and proliferation (Amson et al., 2013). Cell proliferation, migration, and invasion of bladder cancer were restrained by overexpressed miR-218-5p that also promoted the chemosensitivity of bladder cancer to cisplatin by targeting Glut1 (Cheng et al., 2015; Li et al., 2017). Wang et al. (2020) revealed that MNX1-AS1 inhibits bladder cancer cell proliferation, migration, and invasion by manipulated miR-218-RAB1A axis.
Therefore, we speculated whether hsa_circTPT1_003 might be involved in regulating miR-218-5p through the sponge regulation mechanism. We have preliminarily confirmed the speculation by qRT-PCR and bioinformatics analysis. Consistently, using the ENCORI program, we found that CCNE2/SMC4 were positively correlated with miR-218-5p (Fig. 7D). Therefore, we conclude that regulation mechanism of has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 signaling axis might be involved in bladder cancer progression.
Conclusion
In this study, the differential expression profile of circRNAs was screened by using circRNAs microarray in bladder cancer, and the circRNA–miRNA–mRNA network that was likely to be closely associated with mRNA processing or cell cycle was established successfully. And the differential overexpression of circRNAs including hsa_circ_0000133, hsa_circ_0023610, hsa_circ_0005615, hsa_circ_0030162, hsa_circ_0077007, hsa_circ_0001140, and hsa_circ_0107031 was explored between MIBC and NMIBC.
The hsa_circTPT1_003 was selected as a candidate for further research. The expression of hsa_circTPT1_003 was upregulated by qRT-PCR in bladder cancer tissues. The has_circTPT1_003–has-miR-218-5p–CCNE2/SMC4 axis was determined by bioinformatics approaches. The hsa_circTPT1_003-hsa-miR-218-5p-CCNE2–SMC4 axis related to cell cycle was possibly elucidated as a mechanism of tumor progression, which provides a new idea and potential target for effective monitoring and treating bladder cancer patients.
Footnotes
Acknowledgments
We thank TCGA, circBank, dbDEMC 2.0, and miRCancer projects for providing high-quality clinical data on bladder cancer.
Authors' Contributions
J.W. and H.Z. contributed to the conception and design of the research and performed the research, data analysis, and drafted the article. P.W., J.C.S., Y.L., H.Y.D., and J.F. contributed to data analysis. W.P.S., B.J.Q., and Z.J.L. performed the research. J.M.T. contributed to the design of the study. H.Z. and D.W. contributed to the design of the study and revised the article. All authors read and approved the final article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by National Natural Science Foundation of China (Grant No. 82003002), the Natural Science Foundation of Fujian Province (Grant Nos. 2012YZ0001-1 and 2018J01337), Outstanding Youth Cultivation Project of 900 Hospital (Grant No. 2017Q05), and Special Clinical Research of 900th Hospital (Grant No. 2018J18).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.