
Abstract
In this study, we examined the regulatory role of CCDC34 in the sorafenib sensitivity of hepatocellular carcinoma (HCC) and its functional partners. Wide-type Huh7 and Hep3B and induced sorafenib-resistant (SR) Huh7/SR and Hep3B/SR cells were used as in vitro cell models. Immunofluorescent staining and coimmunoprecipitation were performed to check protein–protein interaction. Cell Counting Kit-8 (CCK-8), terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL), PI/Annexin V staining, and western blot analysis were performed to assess cell response to sorafenib. The results showed that CCDC34 upregulation in HCC was associated with poor survival. Huh7/SR and Hep3B/SR cells had significantly higher CCDC34 expression than the parental cell lines. RABL2A expression was significantly upregulated in SR HCC cells and interacted with CCDC34 in its GTP-bound state in Huh7/SR and Hep3B/SR cells. RABL2A depletion sensitized Huh7/SR and Hep3B/SR cells to sorafenib. RABL2A Q80L mutant (GTP-bound state locked), but not S35N mutant (GDP-bound state locked) overexpression increased sorafenib IC50 of Huh7 and Hep3B cells. CCDC34 depletion nearly abrogated the protective effects of RABL2A Q80L overexpression both in vitro and in vivo. RABL2A Q80L overexpression significantly increased the expression of p-p38 and p-JNK, the effects of which were significantly attenuated by CCDC34 depletion. In summary, we infer that the RABL2A–CCDC34 axis plays an important role in mediating p38/MAPK and JNK/MAPK signaling, thereby contributing to acquired sorafenib resistance in HCC.
Introduction
Hepatocellular carcinoma (HCC), is the dominant form of primary liver cancer (Siegel et al., 2017). Although therapeutic strategies have been improved during the past decades, HCC mortality has increased in western countries since only a small proportion of patients were diagnosed at early asymptomatic stages (Zucman-Rossi et al., 2015). Sorafenib is an oral multikinase inhibitor, which is currently used as first-line therapy for advanced HCC. Mechanistically, sorafenib blocks the Raf-MEK-ERK, the vascular endothelial growth factor receptor, and platelet-derived growth factor receptor signaling pathways (Llovet et al., 2008). Although with strong inhibiting effects on multiple critical tumor-driving pathways, sorafenib only prolonged the median overall survival (OS) by 2.3 to 3 months in advanced HCC patients (Cheng et al., 2009). Many HCC patients respond poorly to this drug. A large proportion of patients develop acquired sorafenib resistance after a period of treatment. The underlying mechanisms remain to be elucidated.
The coiled-coil domain-containing (CCDC) is a versatile structural motif in proteins essential for some functional roles, such as regulating gene expression, cell cycle progression, drug metabolism, and membrane fusion. The dysregulation of CCDC proteins is involved in the initiation and malignant transformation of some tumors, such as CCDC170 in breast cancer (Jiang et al., 2017) and CCDC67 in gastric cancer (Park et al., 2012). Coiled-coil domain-containing protein 34 (CCDC34), also called Renal Carcinoma Antigen NY-REN-41, is a protein-coding gene that maps to 11p14.1 in the human genome. Its upregulation is associated with malignant phenotypes of HCC (Lin et al., 2020). However, whether it regulates sorafenib sensitivity in HCC is not clear.
Rab-Like Protein 2A (RABL2A) belongs to the RAS GTPase superfamily, with presumed GTP binding and GTPase activity. Its upregulation is a poor prognostic biomarker in pancreatic cancer (Anand et al., 2020). In its GTP-binding state, RABL2A interacts with intraflagellar transport (IFT)-B complex and participates in ciliary assembly (Nishijima et al., 2017).
In this study, we examined the regulatory role of CCDC34 in sorafenib sensitivity in HCC and its functional partners.
Materials and Methods
Data analysis in The Cancer Genome Atlas (TCGA)-Liver and HCC
Data extraction from TCGA-liver and hepatocellular carcinoma (LIHC) was conducted as previously described (Bai et al., 2019). In brief, only patients with HCC and RNA-seq data were included.
Data mining in HCCDB
CCDC34 expression in HCC and adjacent normal tissues were also analyzed using HCCDB, which archived 15 public HCC gene expression datasets containing over 3900 samples (Lian et al., 2018), from Gene Expression Omnibus (GEO) database, TCGA-LIHC and Liver Cancer-RIKEN, JP Project from International Cancer Genome Consortium (ICGC LIRI-JP).
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed to estimate the biological pathway divergences in TCGA-LIHC cases stratified by the median expression of CCDC34. The software manipulation details and parameter setting were as previously described (Xia et al., 2020).
Cell culture and treatment
HCC cell lines Huh7 and Hep3B were obtained from the National Collection of Authenticated Cell cultures (Shanghai, China). Sorafenib-resistant (SR) Huh7 and Hep3B cells were established according to the method introduced previously (Ewald et al., 2014). In brief, the concentration of sorafenib (Selleckchem, Houston, TX) was gradually increased by 0.25 μM per week. About 7 months later, Huh7/SR and Hep3B/SR cells were established and were continuously maintained in a culture medium with sorafenib.
Lentivirus carrying scramble (scr.) sequence, CCDC34 shRNA, or RABL2A shRNA were constructed with pLKO.1-puro vector. ShRNA sequences are provided in Supplementary Table S1. Lentiviral CCDC34 (NM_080654), lentiviral RABL2A (NM_001306158) expression vector with HA-tag (HA-RABL2A), and the RABL2A Q80L and S35N mutant were generated using pLV-puro vector (Hanbio, Shanghai, China). Lentivirus for infection was produced by cotransfecting the shuttle plasmids, the psPAX2 packaging plasmid, and the pMD2.G envelope plasmid into 293T cells. The lentiviral supernatant was harvested 48 h after transfection.
Reverse transcription quantitative real-time PCR
Total RNAs of cell samples were extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). Quantitative real-time PCR procedures were conducted as described previously (Liu et al., 2020). The primers used for amplification are provided in Supplementary Table S1. GAPDH was used as an internal control.
Western blot analysis
Cells were directly lysed in RIPA buffer (Beyotime, Nantong, China). Then, 30 μg of protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto an nitrocellulose membrane. The membranes were blocked, incubated with primary antibodies at 4°C overnight, washed, and then incubated with secondary antibody conjugated to horseradish peroxidase (HRP) for 1 h. Protein bands were visualized with Chemiluminescent HRP Substrate (EMD Millipore, Billerica, MA). Primary antibodies used include anti-CCDC34 (1:500, DF13697; Affinity Biosciences, Jiangsu, China), anti-RABL2A (1:500, 17816-1-AP; Proteintech, Changzhou, China), anti-caspase-3 (1:1000, 19677-1-AP; Proteintech), anti-PARP (1:1000, 13371-1-AP; Proteintech), anti-HA-tag (1:3000, 51064-2-AP; Proteintech), anti-Flag-tag (1:500, 20543-1-AP; Proteintech), anti-p-p38 (Thr180/Tyr182, 1:1000, AF4001; Affinity Biosciences) anti-p38 (1:1000, 14064-1-AP; Proteintech), anti-p-JNK (Thr183/Tyr185, 1:1000, AF3318; Affinity Biosciences), and anti-JNK (1: 300, 51151-1-AP; Proteintech).
Cell Counting Kit-8 and colony formation assay of cell viability
Huh7/SR and Hep3B/SR cells with CCDC34 knockdown or RABL2A overexpression were seeded in 96-well plates at a density of 5000 cells/well. Twenty-four hours later, cells were treated with different concentrations of sorafenib for 48 h. Cell viability was measured using the Cell Counting Kit-8 (CCK-8; Beyotime) according to the manufacturer's instructions. Colony formation assay was performed in six-well culture plates, with 1000–2000 cells/well. Then the cells were incubated for 2 weeks, with or without the presence of sorafenib. Then, colonies were fixed in 4% paraformaldehyde and stained with 1% Crystal Violet. The number of colonies was counted, and the relative colony formation efficiency (%) was calculated.
Terminal deoxynucleotidyl transferase-mediated nick end labeling staining and flow cytometric analysis of cell apoptosis
Huh7/SR and Hep3B/SR cells with or without CCDC34 knockdown were treated with sorafenib for 48 h. Apoptotic cells were detected by terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay staining (TUNEL Assay Kit-FITC, ab66108; Abcam, Cambridge, UK) or FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions. Flow cytometric analysis was performed using the BD FACSCanto II flow cytometer (BD Biosciences) instrument.
Immunofluorescent staining
Huh7/SR and Hep3B/SR cells were grown on coverslips, fixed with ice-cold methanol, permeabilized with 0.5% (v/v) Triton X-100 in phosphate-buffered saline (PBS), and incubated with blocking buffer. Then, coverslips were incubated with a primary antibody against human CCDC34 (DF13697) for 1 h at 37°C, washed with PBST (0.2% Tween 20 in PBS), and subsequently incubated with a secondary antibody [anti-rabbit IgG (H+L), F(ab’)2 Fragment, and Alexa Fluor® 647 Conjugate] (Cell Signaling, Danvers, MA). Then, coverslips were washed thoroughly and incubated with a primary antibody against human RABL2A (MA5-25314; Thermo Fisher, Franklin, MA) for 1 h at 37°C, washed with PBST (0.2% Tween 20 in PBS), and subsequently incubated with goat Anti-Mouse IgG H&L conjugated with Alexa Fluor 488 (ab150113; Abcam) for 30 min at room temperature. Then, the coverslips were washed and mounted with fluorescent mounting medium (Agilent, Santa Clara, CA). Immunofluorescent (IF) images were captured using a fluorescence microscopy (Nikon, 90i, Japan).
Coimmunoprecipitation
Huh7/SR and Hep3B/SR cells were infected with lentivirus for Flag-CCDC34 and HA-RABL2A overexpression. Forty-eight hours later, cells were harvested and lysed with RIPA buffer. To check the interaction among endogenous proteins, MG132 was added 6 h before harvesting the samples. The lysates were centrifuged to collect supernatant and then was precleaned by Pierce protein A/G PLUS-Agarose (Thermo Scientific, Waltham, MA). After that, IP was performed using anti-Flag-tag, anti-HA-tag, anti-CCDC34, or anti-RABL2A at 4°C for 6 h under gentle agitation. Then, protein A/G PLUS-Agarose beads were added to the mixture, with rotary agitation for 4 h at 4°C. The immunoprecipitated complexes were collected, washed, and subjected to western blot analysis. The input was used as a positive control.
Animal studies
All procedures involving animals were approved by the Sichuan Provincial People's Hospital, China (approval no. 2017-126-7). Ninety 6-week-old male BALB/c-nu mice were purchased from the Dossy Experimental Animals (Chengdu, China). Mice were housed in a Specific Pathogen-Free facility. Mice were randomly divided into three individual tests (n = 30 per test), with five groups (n = 6 per group) in each test. Twenty-four hours after lentiviral infection, Huh7 cells [1 × 106 in 200 μL DMEM:Matrigel (1:1 ratio)] were implanted into the left flanks of the mice. Mice in group 1 were only injected with Huh7 cells infected with vector control. Group 2 to 5 received sorafenib treatment (25 mg/kg, daily through oral gavage) when tumors reached ∼50 mm3, until the termination of the experiment. Tumor dimensions were recorded every 3 days using a caliper. Tumor volumes were calculated as follows: volume = d2*D/2, where d and D is the shortest and longest tumor diameter, respectively. All mice were sacrificed on day 43 since tumor cell inoculation. Tumors were removed and photographed. Then, the tumors were sectioned for Hematoxylin and Eosin staining, Immunohistochemistry staining of Ki-67, and TUNEL staining.
Statistical analysis
Data analysis was performed using GraphPad Prism 8.10 (GraphPad, Inc., La Jolla, CA). For multiple group comparison, one-way ANOVA with post hoc Tukey's multiple comparisons test was conducted. For two-group comparison, Welch's unequal variances t-test was performed to detect the differences. Kaplan–Meier survival curves were generated by the median gene expression. Log-rank test was performed to determine the significance of the difference between the survival curves. p < 0.05 was considered to be statistically significant, with * and
Results
CCDC34 upregulation is associated with sorafenib resistance in HCC
In HCCDB (Lian et al., 2018), 10 databases had CCDC34 expression data from both HCC and adjacent normal tissues. CCDC34 expression was upregulated in HCC than in adjacent normal tissues in 9/10 of the datasets (Supplementary Fig. S1A). CCDC34 IHC staining data from the Human Protein Atlas (HPA) (Uhlen et al., 2010) confirmed CCDC34 expression at the protein level, mainly in the nuclear part of HCC cells (Supplementary Fig. S1B). High CCDC34 expression was associated with significantly worse OS and progression-free survival in TCGA-LIHC (Supplementary Fig. S1C, D) and was associated with significantly worse OS in ICGC-LIRI-JP dataset (Supplementary Fig. S1E).
To explore the functional relevance of CCDC34 in HCC, we conducted single-gene GSEA (median CCDC34 separation) analysis using TCGA-LIHC (Supplementary Fig. S1F, top panel). The high CCDC34 group has significantly upregulated genes enriched in DNA Repair, E2F Targets, G2M Checkpoint, Mitotic Spindle, Spermatogenesis, and MYC Targets V1 (Supplementary Fig. S1F, bottom panel). As DNA repair, G2M Checkpoint, and Mitotic Spindle are closely linked to the chemosensitivity of cancer cells, we generated SR Huh7 and Hep3B cells (Supplementary Fig. S1G), which had significantly higher CCDC34 expression than the wild-type (WT) parental cell lines (Supplementary Fig. S1H, I).
CCDC34 expression is vital for sustaining sorafenib resistance in HCC
To explore the regulatory function of CCDC34 in sorafenib resistance, Huh7/SR and Hep3B/SR cells were subjected to lentivirus-mediated CCDC34 knockdown (Fig. 1A, B). CCDC34 knockdown significantly reduced the viability of Huh7/SR and Hep3B/SR cells (Fig. 1C–E). Besides, it sensitized cells to the sorafenib-induced cell growth arrest (Fig. 1C–E) and cell apoptosis (Fig. 1F–H).
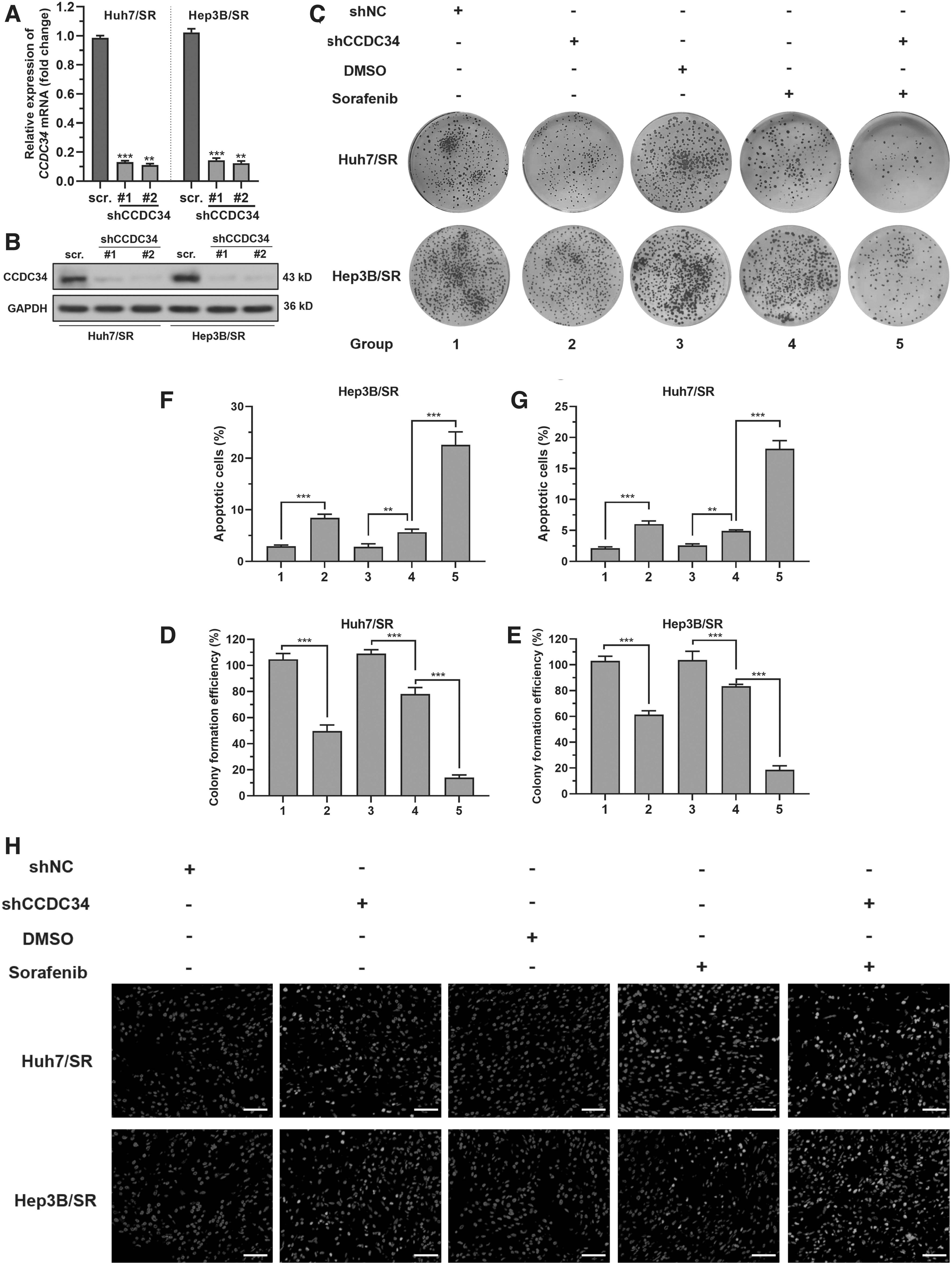
CCDC34 expression is vital for sustaining sorafenib resistance in HCC.
RABL2A interacts in its GTP-bound state with CCDC34 in HCC cells
Bioinformatic prediction showed that RABL2A is a high potential interactor of CCDC34 (Fig. 2A). RABL2A is also upregulated in Huh7/SR and Hep3B/SR cells than in the parental cell lines (Fig. 2B). IF staining showed that RABL2A and CCDC34 only had weak colocalization in WT Huh7 and Hep3B cells (Fig. 2C, top panel). However, colocalization was significantly enhanced near the nuclear membrane area in Huh7/SR and Hep3B/SR cells (Fig. 2C, bottom panel). Coimmunoprecipitation (Co-IP) assay confirmed the interaction between HA-RABL2A and Flag-CCDC34 Huh7/SR and Hep3B/SR cells (Fig. 2D). When MG132 was added, the interaction between endogenous RABL2A and CCDC34 was confirmed (Fig. 2E). Then we constructed the HA-tagged RABL2A Q80L mutant that locked in a GTP-bound state, and the RABL2A S35N mutant, which is a GDP-bound form (Nishijima et al., 2017). Results showed that the Q80L mutant, but not S35N mutant, interacted with CCDC34 (Fig. 2F).
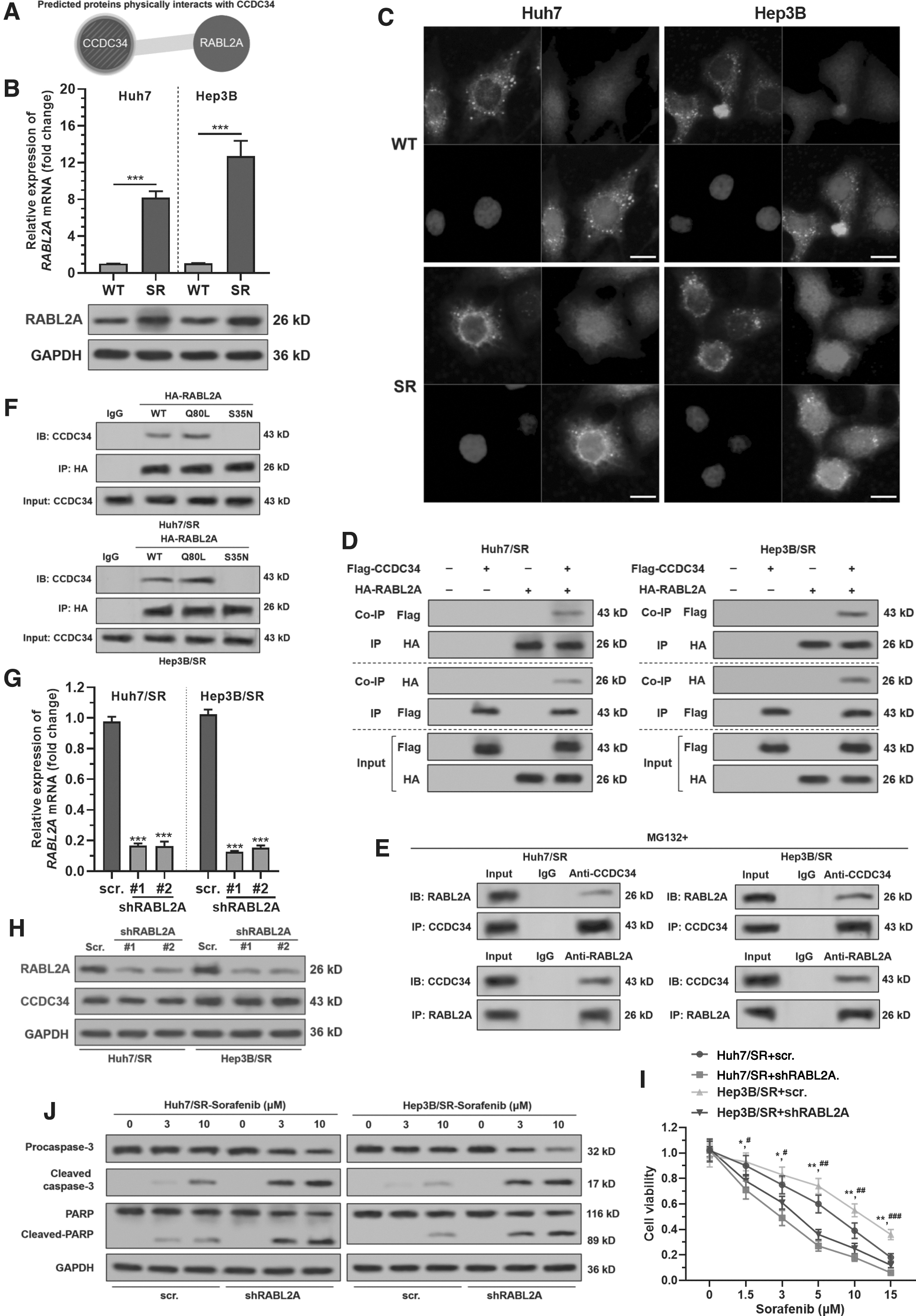
RABL2A interacts in GTP-bound state with CCDC34 in SR HCC cells.
RABL2A depletion increases sorafenib sensitivity of HCC cells
To explore whether RABL2A regulates sorafenib sensitivity, Huh7/SR and Hep3B/SR cells were subjected to lentivirus-mediated RABL2A knockdown (Fig. 2G, H). RABL2A knockdown did not alter CCDC34 expression at the protein level (Fig. 2H).
RABL2A depletion significantly reduced the cell viability of Huh7/SR and Hep3B/SR cells after sorafenib treatment (Fig. 2I) and significantly increased the expression of apoptosis-associated cleaved caspase-3 and cleaved PARP (Fig. 2J).
RABL2A in its GTP-bound state promotes sorafenib resistance by CCDC34-mediated p38/JNK MAPK signaling
To verify the contribution of RABL2A GDP-bound or GTP-bound form to sorafenib resistance, WT Huh7 and Hep3B cells were subjected to lentivirus-mediated RABL2A Q80L or S35N mutant overexpression (Fig. 3A, B). RABL2A Q80L, but not S35N mutant overexpression significantly increased the IC50 to sorafenib (Fig. 3C, D), and weakened the inhibitive effect of sorafenib on colony formation (Fig. 3E). CCDC34 knockdown partially canceled the protective effects of RABL2A Q80L overexpression on sorafenib-induced cell growth arrest (Fig. 3E), cell apoptosis (Fig. 3F, G), and the expression of apoptosis-related proteins (Fig. 3H).
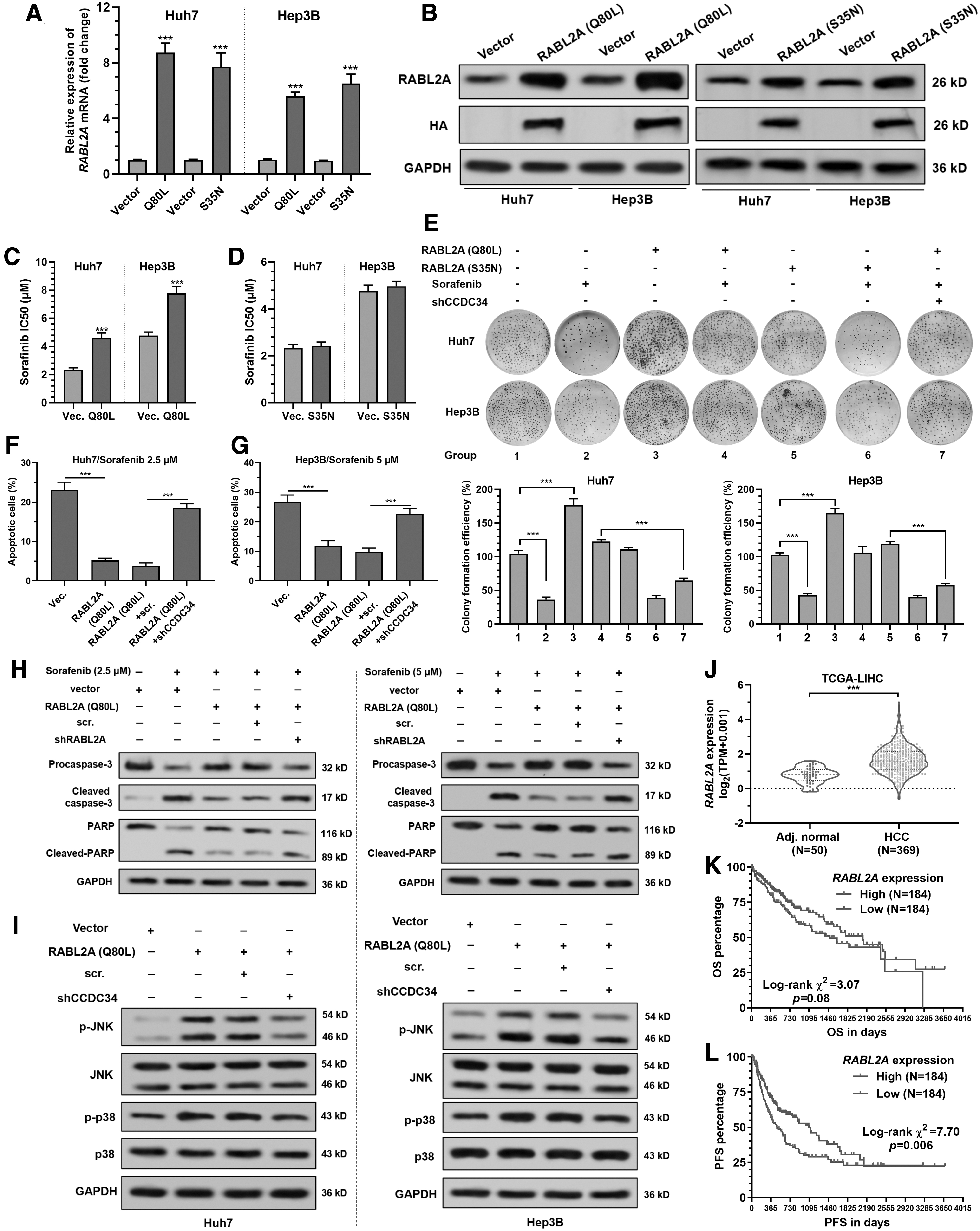
RABL2A promotes sorafenib resistance by CCDC34-mediated p38/JNK MAPK signaling.
In both Huh7 and Hep3B cells, RABL2A Q80L overexpression significantly increased the expression of p-p38 and p-JNK (Fig. 3I), suggesting that it might activate some of the MAPK signaling pathways. CCDC34 depletion significantly attenuated RABL2A Q80L-induced p-p38 and p-JNK expression (Fig. 3I).
Data from TCGA-LIHC showed that RABL2A expression is significantly upregulated in HCC (Fig. 3J) and was associated with significantly worse survival outcomes (Fig. 3K–L).
RABL2A promotes sorafenib resistance by CCDC34 in vivo
To verify whether the RABL2A–CCDC34 axis modulates sorafenib resistance in vivo, we generated xenograft tumor models in male BALB/c-nu mice based on Huh7 cells. The results indicated that cells with RABL2A (Q80L) overexpression had earlier tumor formation (∼15 days after inoculation) (Fig. 4B). Sorafenib treatment significantly suppressed tumor cell growth but enhanced apoptosis and necrosis (Fig. 4A–C). RABL2A Q80L, but not S35N mutant overexpression, canceled sorafenib-induced growth suppression, apoptosis, and necrosis (Fig. 4A–C). CCDC34 knockdown abrogated the protective effects of RABL2A Q80L overexpression (Fig. 4A–C).

RABL2A promotes sorafenib resistance by CCDC34 in vivo.
Discussion
CCDC34 has oncogenic properties in some cancers, through activating angiogenetic processes (Hu et al., 2018) and the MAPK and AKT pathways (Gong et al., 2015). In HCC, CCDC34 upregulation contributes to poor survival by promoting proliferation and metastasis of tumor cells (Lin et al., 2020). However, its pathophysiological function in HCC remains poorly understood.
Our GSEA results showed that the high CCDC34 expression group had upregulated gene-enriched multiple gene sets related to HCC pathogenesis, such as DNA Repair, E2F Targets, G2M Checkpoint, Mitotic Spindle, and MYC Targets V1. These mechanisms might partly explain the association between CCDC34 expression and poor survival of patients with HCC. Besides, they also imply that CCDC34 expression might be associated with the chemosensitivity of HCC. Using induced Huh7/SR and Hep3B/SR as in vitro cell models, we observed that CCDC34 knockdown increased sorafenib sensitivity. Therefore, CCDC34 might act as a modulator of cellular responses to sorafenib.
Bioinformatic and Co-IP results indicated that RABL2A in its GTP-bound state physically interacts with CCDC34 SR HCC cells. RABL2A depletion significantly sensitized Huh7/SR and Hep3B/SR cells to sorafenib. RABL2A Q80L mutant, but not S35N mutant overexpression increased sorafenib IC50 of Huh7 and Hep3B cells. CCDC34 depletion nearly abrogated the protective effects of RABL2A Q80L overexpression. These findings suggested that CCDC34 might act as a downstream signaling mediator of RABL2A in promoting sorafenib resistance.
RABL2 interacts with CEP350 and FOP and directs the ciliary entry of intraflagellar transport (IFT) protein, which is responsible for the trafficking of signaling molecules (Kanie et al., 2017). It also binds to the IFT74-IFT81 heterodimer in its GTP-bound state and participates in ciliary/flagellar assembly (Nishijima et al., 2017; Duan et al., 2021). Generally, when bound to GTP, Ras GTPase is active and can activate downstream effector proteins (Nan et al., 2015). Therefore, we infer that the GTP-bound state might be important for the oncogenic role of RABL2A.
In bladder cancer, CCDC34 upregulation was associated with augmented MAPK signaling, including ERK/MAPK, p38/MAPK, and JNK/MAPK (Gong et al., 2015), suggesting that CCDC34 might act as an important mediator of these signaling pathways. In the current study, we found that RABL2A did not alter CCDC34 protein expression. However, RABL2A Q80L activates the expression of p-p38 and p-JNK through CCDC34. The p38/MAPK and JNK/MAPK signaling pathways can control the balance of apoptosis and autophagy in response to chemotherapeutic agents, depending on cell type, stress signal, and other circumstances (Sui et al., 2014; Yao et al., 2020). In HCC, sorafenib-induced autophagy might serve as a prosurvival mechanism (Zhai et al., 2014; Tong et al., 2018). p38α knockdown (by shRNAs or pharmacologic compounds), sensitizes mouse HCC to sorafenib therapy (Rudalska et al., 2014). Activation of JNK can predict a poor response to sorafenib in HCC (Hagiwara et al., 2012). JNK inhibitor also eliminates sorafenib resistance (Nguyen et al., 2014).
In summary, we infer that the RABL2A–CCDC34 axis plays an important role in mediating p38/MAPK and JNK/MAPK signaling, thereby contributing to acquired sorafenib resistance in HCC.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
Chengdu Science and Technology Innovation research and development project 2021-YF05-02143-SN and the Sichuan Science and Technology Office (No. 3050410336).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.