
Abstract
Hereditary sensory neuropathy type 1A (HSN1A) is an autosomal, dominantly inherited peripheral neuropathy caused by mutations in serine palmitoyl transferase long chain 1 (SPTLC1), involved in the de novo synthesis of sphingolipids. We have previously reported calcium imbalance, as well as mitochondrial and ER stress in both HSN1 patient lymphoblasts and a transiently transfected cell model. In this study, we investigated the role of the Ca2+-activated protease calpain in destabilizing the cell cytoskeleton, by examining calpain activity in SH-SY5Y cells overexpressing the V144D mutant and changes in microtubule-associated proteins (MAP). Intramitochondrial Ca2+ was found to be significantly depleted and cytoplasmic Ca2+ increased in the V144D mutant. Subsequently, calpain and proteasome activity were increased and calpain substrates, microtubule associated proteins MAP2, and tau were significantly reduced in the microtubule fraction of the mutant. Significant changes were also found in motor proteins dynein and KIF2A detected in the microtubule fraction of cells overexpressing the V144D mutation. There was also a reduction in anterograde and retrograde mitochondrial transport velocities in the V144D mutant. These findings strongly implicate cytoskeletal aberration caused by Ca2+ dysregulation and subsequent loss of microtubule transport functions as the cause of axonal dying back that is characteristic of HSN1.
Introduction
Hereditary sensory neuropathy type 1 (HSN1) is one of the most common sensory neuropathies displaying a progressive loss of pain and temperature sensation in the distal limbs, with distal and later proximal limb weakness (Nicholson, 2002; Houlden et al., 2004, 2005). Owing to loss of sensation, painless injuries develop, often complicated by ulcerations and osteomyelitis, eventually requiring amputation (Nicholson, 2002). There are seven known mutations in the SPTLC1 gene that result in HSN1: p.V144D, p.C133W, p.C133Y, p.C133R, p.S331F, p.S331Y, and p.A352V (Dawkins et al., 2001; Rotthier et al., 2009, 2011; Auer-Grumbach et al., 2013).
SPTLC1 forms a subunit of the serine palmitoyl transferase (SPT) holoenzyme, which is localized in the outer membrane of the endoplasmic reticulum and catalyses the rate-limiting step in sphingolipid synthesis. Mutations in SPTLC1 cause a shift in substrate specificity of SPT from
Ca2+ dysregulation in ER and mitochondria has previously been documented in HSN1 patient lymphoblasts (Myers et al., 2014), DRG cultures (Wilson et al., 2018), and ND15 cell model containing the V144D mutation (Stimpson et al., 2016). A consequent triggering of Ca2+-activated protease systems in these cells would thus seem quite likely.
Calpains are Ca2+-dependent cysteine proteases that can be activated by micro- (calpain I) or millimolar (calpain II) levels of Ca2+, and these activities are tightly regulated by the endogenous calpain inhibitor, calpastatin (Hanna et al., 2008; Rao et al., 2008). Calpains recognize and cleave various cytoskeletal proteins including actin-binding proteins (e.g., spectrin, actinin, talin, and ankyrin), microtubule-binding proteins (e.g., MAP2 and tau), and neurofilaments among others (Johnson et al., 1991; Fifre et al., 2005; Camins et al., 2006; Lebart and Benyamin, 2006).
Microtubules are stabilized by microtubule-associated proteins (MAPs), which work as cargo tracks along axons, facilitating transport of materials along the length of the axon, with the help of specific motor proteins. As a whole, microtubules and associated proteins play an important role in maintaining the functional integrity of neurons. Microtubule impairment and subsequent disruption in cargo transport has been widely implicated in a number of neurodegenerative disorders including amyotrophic lateral sclerosis (Baird, 2013), prion disease (Guo et al., 2012), frontotemporal dementia, and numerous tauopathies (Xie and Miyasaka, 2016).
The implication of this would be particularly significant in peripheral neurons where axonal length varies from a few millimeters to up to a meter, making axonal transport a pivotal process of cell survival and functionality. Although many studies have looked at abnormal Ca2+ handling in HSN1 cells, none have directly linked Ca2+ dysregulation with cytoskeletal abnormalities. Here we tested the hypothesis of a Ca2+-activated pathway that links HSN1 mutation to calpain activation and the consequential impact on calpain substrates, the microtubule stabilizing proteins. The data strongly implicate cytoskeletal alterations and the loss of microtubule transport functions as the cause of the axonal dying back characteristic of HSN1.
Materials and Methods
All cell culture consumables were purchased from Greiner (Interpath) and chemicals from Sigma-Aldrich unless specified otherwise. SH-SY5Y neuroblastoma cells were purchased from Sigma-Aldrich (ECACC). All cell culture media and supplements including Lipofectamine 3000, Geneticin, Rhod3-AM, and Mitotracker-Red were purchased from GIBCO Invitrogen, Thermo-Fisher. Paclitaxel, GTP, Rhod-2AM, Mitochondrial Cytopainter—Blue, proteasome inhibitor, and calpain inhibitor were purchased from Abcam. Calpain-Glo™ Protease Assay kit was purchased from Promega Australia.
Generation of stably transfected cell lines
SH-SY5Y cells were cultured in Dulbecco's modified Eagle's/F12 medium with 10% fetal bovine serum (SFBS; Bovogen), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM
Detection of cytoplasmic and mitochondrial calcium
Cytoplasmic and mitochondrial calcium was assessed by labeling the cells with Rhod-3AM and dihydro-rhod-2AM, respectively, and analyzed using LSM800 (Zeiss) confocal microscope. Methods are described in detail in Supplementary Data S1.
Calpain and proteasome activity
Protease activity was measured using the Calpain-Glo Protease Assay kit, in cells seeded in 96-well white plates. Calpain activity was measured in cells pretreated with chymotrypsin-proteasome inhibitor MG132 (4 μM) for 18 h and proteasome activity was measured in cells pretreated with calpain inhibitor MDL28170 (75 μM) for 30 min in media containing 1% FBS. Reagent preparation, addition, and detection were performed according to manufacturer's instructions and luminescence was measured using a BMG-Polarstar Plate Reader. The measured calpain and proteasome activity was normalized to the amount of protein per well and expressed as luminescence units per mg protein. Sample size was as follows: n = 8 wells per group.
Cell lysis and protein detection
For whole cell lysate protein detection, control and transfected cells were harvested and extracted in NDRM buffer (10 mM Tris pH 8.0, 150 mM NaCl, 1% Triton-X, 1X protease inhibitors) and 50 μg protein was loaded per lane on 8% polyacrylamide gels for the resolution of MAP2 and spectrin α II, and 12% polyacrylamide gels for the resolution of all other cytosolic proteins. 25 μg protein was loaded per lane for MAPs. After imaging in LAS 4000 Image Reader, blots were stripped and reprobed for GAPDH (for total cytosolic fractions) or β-3-tubulin (for microtubule fractions). Band intensities for each protein were quantified using ImageJ and normalized against corresponding GAPDH or β-3-tubulin intensities, and fold change relative to control calculated. All calculations were based on n = 3 immunoblots.
Microtubule isolation and detection of microtubule-bound proteins
Microtubules and microtubule-binding proteins were isolated as described elsewhere (Sloboda, 2015). Approximately 6 × 107 cells (eight T75 flasks) per group were used to isolate microtubule fraction. Pelleted microtubules were used for western blotting and immunofluorescent staining. For western blotting, 25 μg protein was loaded per well to detect levels of MAP2, tau, KIF2A, and dynein bound to microtubule assembly. After imaging, the blots were stripped and reprobed for β-3-tubulin. All calculations were based on n = 3 immunoblots.
Tracking mitochondrial transport
Cells were differentiated in media containing 1% FBS and 10 μM retinoic acid for 7 days, with fresh media supplied every 2 days. On the day of the experiment, cells were treated with 50 nM Mitotracker-Red for 30 min in serum-free media, after which they were washed and loaded with HBSS before image acquisition. Only neurites 50 μm or longer were selected for measuring transport. Images were taken in a time series mode, using a Leica TCS SP5 laser scanning confocal microscope with a 100 × objective, with a scan performed every 5 s for 60 frames, using Zen software (version 2.3).
Experimental design and statistical analysis
Data are expressed as fold change or mean ± SEM, unless specified otherwise. For multiple comparisons, a one-way analysis of variance (ANOVA) was performed, unless mentioned otherwise, using GraphPad Prism version 4.03 software, with Tukey's multiple comparison test. A value of p ≤ 0.05 was considered significant.
Results
Expression of stably transfected SPTLC1 wild-type and V144D mutants
Expression of stably transfected SPTLC was confirmed by immunodetection of both endogenous and transfected SPTLC1 as well as GFP using SDS-PAGE-western blotting. Endogenous SPTLC1, detected at 55 kDa, showed no changes in expression between the nontransfected (NT) and transfected groups (Fig. 1A, C). GFP-tagged SPTLC1 was detected in WT and V144D cell lines at ∼80 kDa (Fig. 1A, C). GFP was also detected in WT and V144D mutants at 80 kDa (Fig. 1B, C), confirming the establishment of stable WT and V144D mutant cell lines.
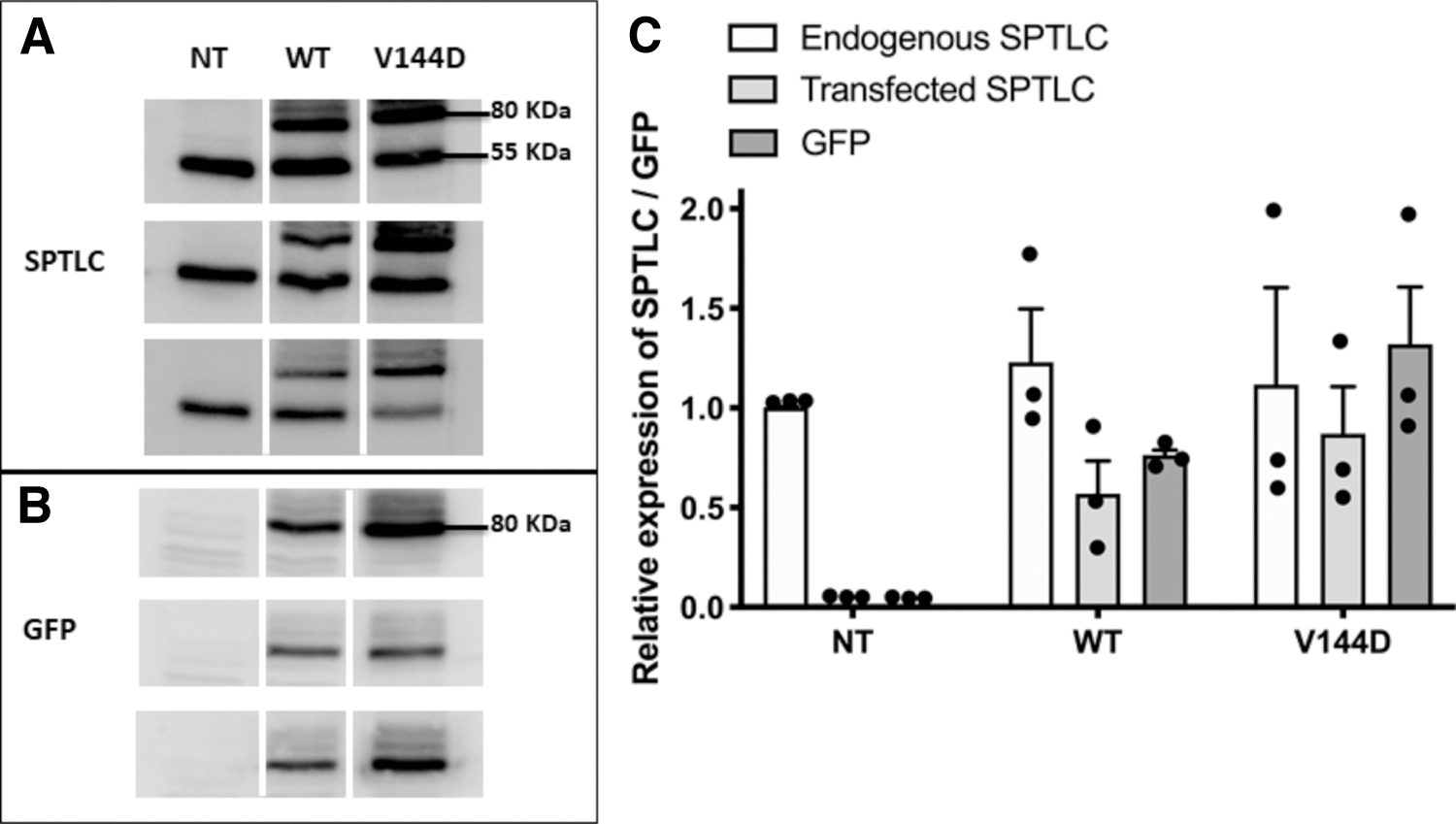
Establishment of stable cell lines. SH-SY5Y cells were transfected with vectors for GFP-tagged SPTLC wild-type (WT) and V144D vectors and selected using Geneticin. Control cells are designated as non-transfected (NT). Cells were harvested, lysed, and protein expression confirmed by immunoblotting using antibodies specific for SPTLC
Cytosolic and mitochondrial Ca2+ dysregulation in V144D mutants
Using the indicator Rhod-3AM, cytosolic Ca2+ showed an increase in WT (1.7-fold) and V144D mutants (2.5-fold) compared with the NT cells (Fig. 2A, B). A slight but significant decrease in mitochondrial Ca2+ was observed in the V144D mutants, as detected by dihydro-Rhod-2AM (Fig. 2C, D). Dihydro-Rhod-2AM was also colocalized with the mitochondrial stain Cytopainter confirming that the calcium detected by dihydro-Rhod-2AM was localized within mitochondria (Fig. 2E). Overall, we detected over 100% increase in cytoplasmic Ca2+ and a 30% decrease in mitochondrial Ca2+ in V144D mutants compared with the NT cells.
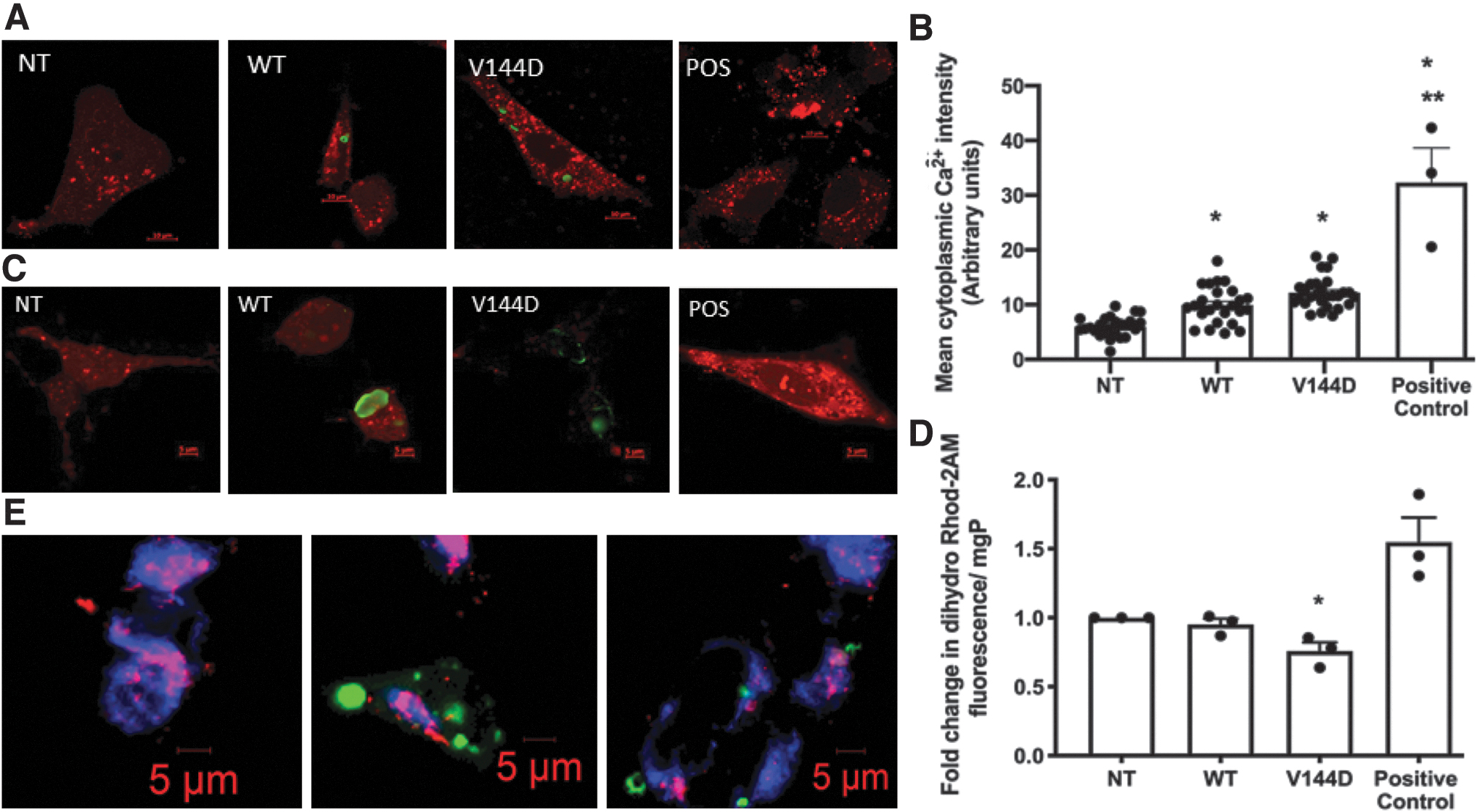
Measurement of relative cytosolic and mitochondrial Ca2+ levels. Cytosolic Ca2+ was measured using Rhod-3AM and mitochondrial Ca2+ using dihydro-Rhod-2AM in cells seeded in 35 mm glass bottom dishes or in black-walled 96-well plates or both. Cells treated with 2 μM ionomycin were used as positive controls. For Rhod-3AM, images were acquired using an LSM800 confocal microscope and mean intensity per cell was calculated in all groups. Representative confocal micrographs of cytosolic Ca2+ staining (red) in NT, WT, V144D, and positive control cells are provided in
V144D mutants show significantly increased calpain and proteasom e activity
We have previously reported that the V144D mutation increases intracellular Ca2+ loading in ND15 cells where a twofold increase in cytoplasmic Ca2+ concentration was detected (Stimpson et al., 2016). We therefore investigated whether Ca2+-dependent protease systems—calpain and the proteasome—are activated in the V144D mutant cells. Calpain (Fig. 3A) and proteasome (Fig. 3B) activities were significantly increased in the V144D mutant compared with the NT and WT cells. V144D showed a 2.7-fold increase in calpain activity and 1.8-fold increase in proteasome activity compared with NT cells, and these activities were also increased significantly in the WT cells, but to a lesser extent than in the V144D cells.
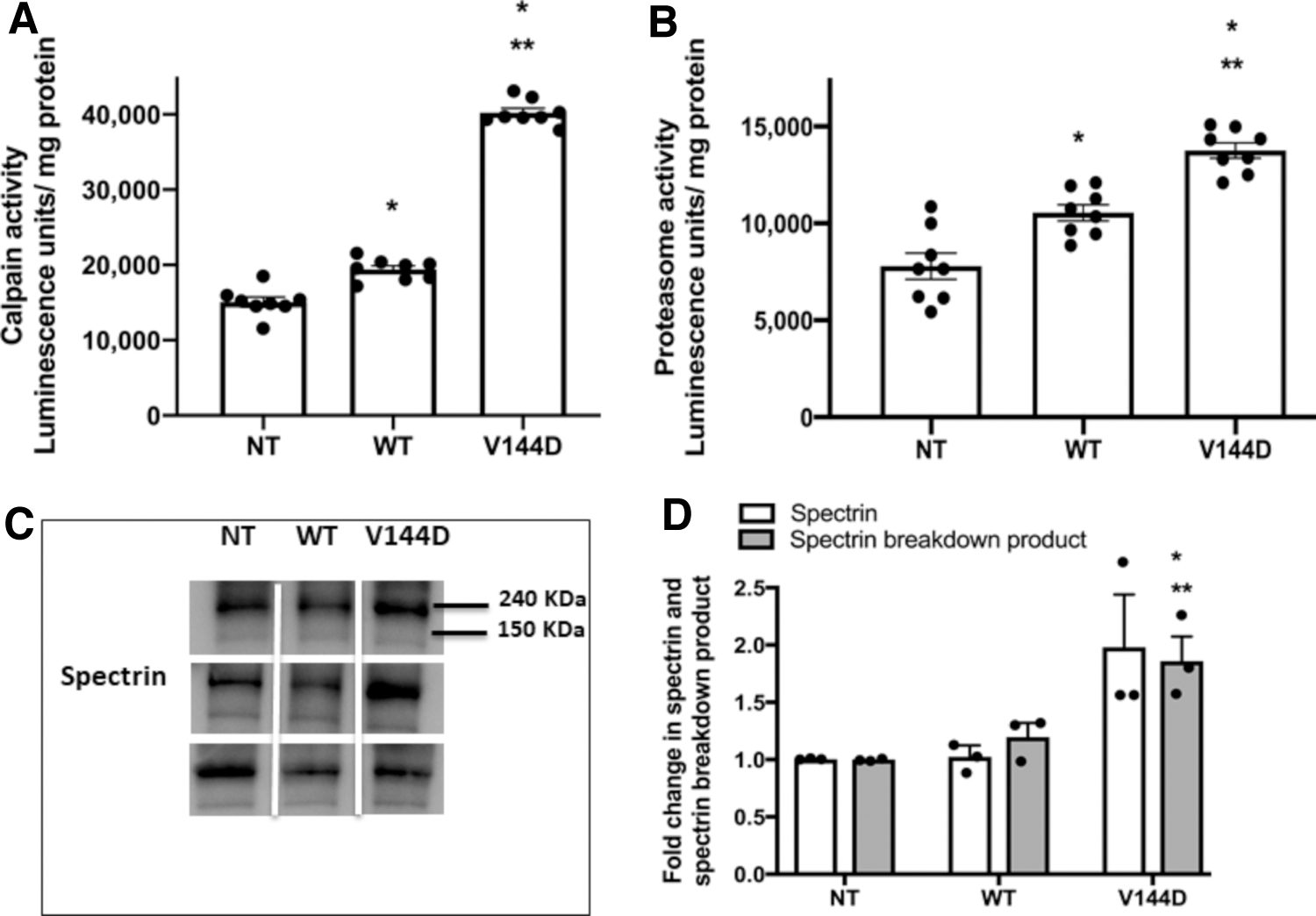
Analysis of protease activity. Stably transfected SH-SY5Y cells were grown in white 96-well plates and calpain
The increase in calpain activity was further confirmed by assessing the presence of spectrin α II breakdown products (SBP) in total cell lysate. Activated calpain cleaves the actin-bundling protein spectrin α II (240 kDa) to a 150 kDa fragment. There was a 1.8-fold increase in SBP in V144D mutants compared with NT, indicating increased calpain activity and consequently increased proteolysis of spectrin α II, yielding SBP (Fig. 3C, D).
Decreased microtubule binding of MAP2 and tau in V144D mutants
Microtubule-bound MAP2 and tau isolated using paclitaxel was examined. Overall, the presence of MAP2 in total cell lysate was found to be significantly reduced in V144D mutants compared with the controls (Fig. 4A, B and Supplementary Fig. S1A). Microtubule-bound-MAP2 was also significantly reduced in V144D compared with NT and WT groups (Fig. 4A, B). This decrease was also reflected in immunostaining of isolated microtubules (Supplementary Fig. S1C), in which NT and WT showed more intensely stained MAP2 compared with the V144D mutant group.
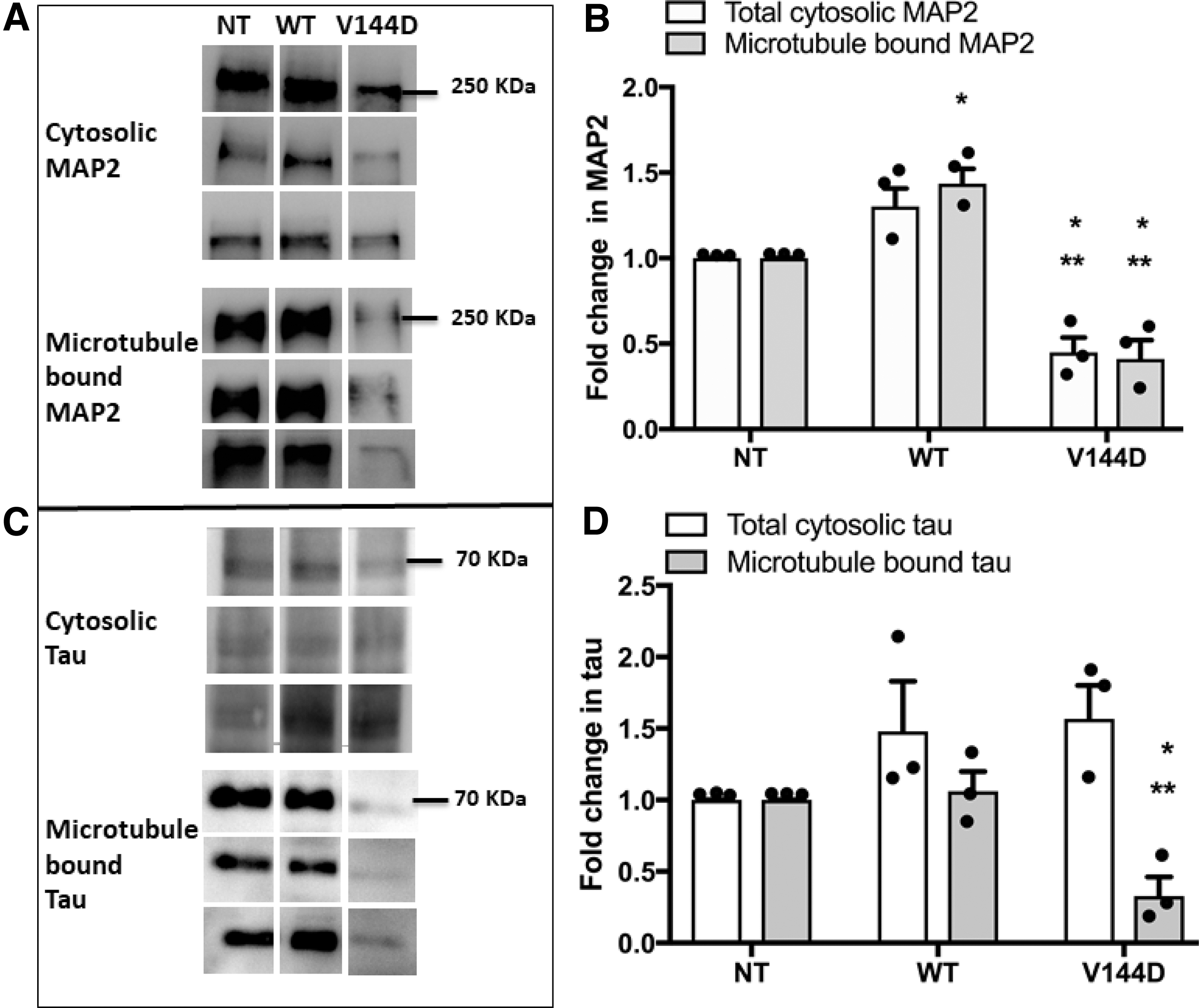
Analysis of expression and microtubule binding of microtubule-associated proteins MAP2 and tau. Presence of MAP2
Expression of MAP tau appeared to be modestly increased in the transfected cells compared with the NT, although the change was not statistically significant (Fig. 4C, D and Supplementary Fig. S1B). However, similar to MAP2, microtubule-bound tau was found to be significantly decreased in V144D compared with the controls NT and WT (Fig. 4C, D). Reduction in microtubule bound tau was also observed in isolated microtubules cryostained for tau, in which the isolate from the V144D mutant cells had regions of less intensely stained tau compared with the control groups (Supplementary Fig. S1D).
Altered microtubule binding of motor proteins in V144D mutants
Microtubule-bound KIF2A and dynein, responsible for microtubule disassembly and retrograde axonal transport, respectively, were examined. Although the total cytosolic KIF2A remained unchanged among the groups, KIF2A was found to be significantly reduced in the microtubule fraction from V144D mutants (Fig. 5A, B).
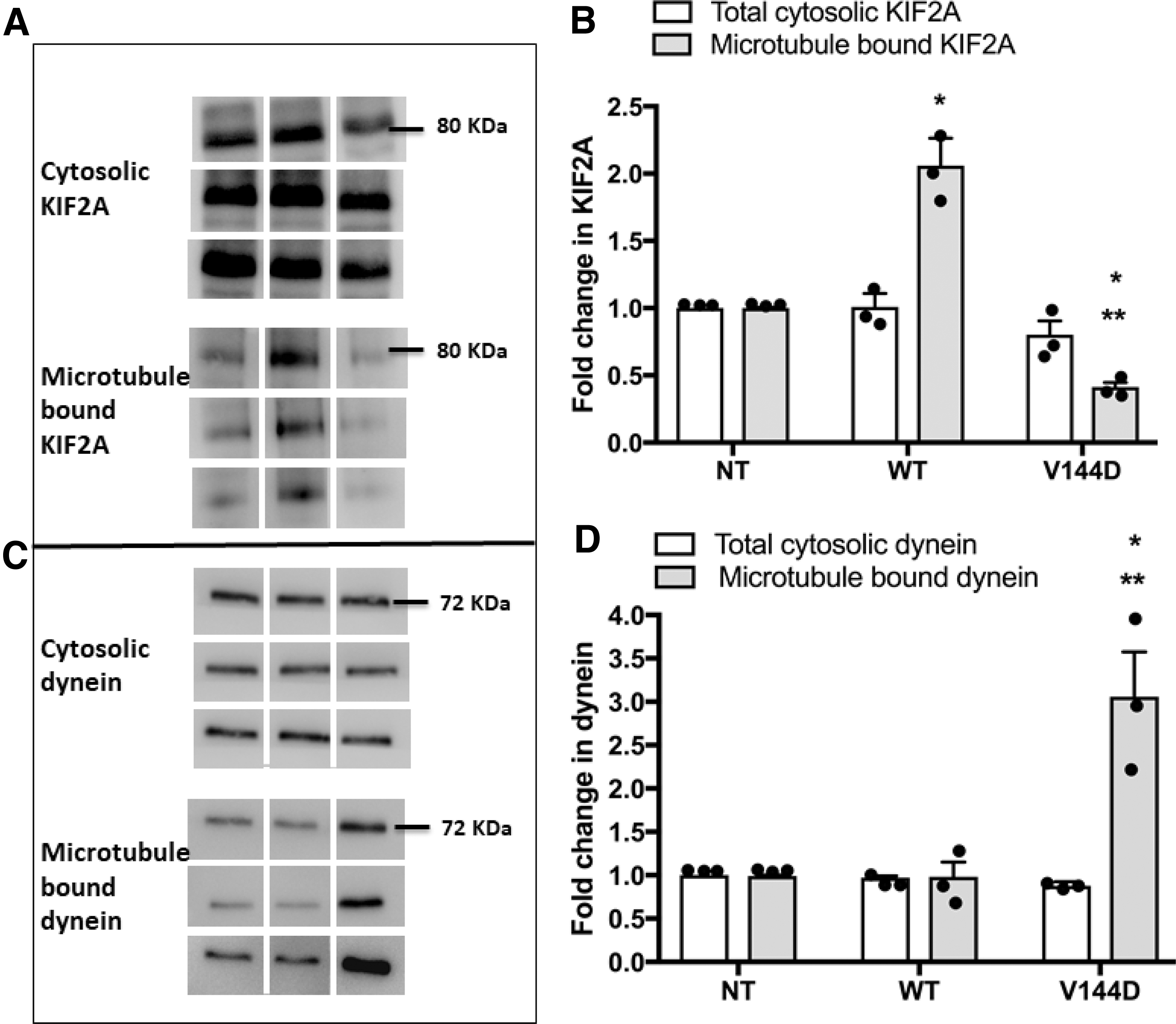
Analysis of expression and microtubule binding of KIF2A and dynein. Presence of KIF2A
Isolated microtubules also showed a decrease in KIF2A cryostaining in V144D mutants compared with the controls (Supplementary Fig. S2A). Of interest, WT cells showed a significant increase in microtubule-bound KIF2A. Dynein in the microtubule fractions appeared significantly higher in the V144D mutants than in the control groups (Fig. 5C, D). Dynein staining in cryosections of V144D microtubules also appeared much brighter relative to the controls (Supplementary Fig. S2B). Nonetheless, the level of dynein in total cell lysate remained unchanged.
Decreased mitochondrial transport velocity in V144D mutants
Mitochondrial movement was tracked by staining the cells using Mitotracker-Red and performing a time series study (Supplementary Fig. S3). Average mitochondrial velocity in both anterograde and retrograde directions were significantly reduced in V144D mutants compared with the controls (Fig. 6A–D). Overall, average retrograde velocity was found to be higher, with mitochondrial transport reaching velocities as high as 4 μm/s in both directions. Average anterograde velocity in control cells was found to be 1.2 μm/s in both NT (Fig. 6A) and WT (Fig. 6B), whereas it was three times slower (0.34 μm/s) in V144D mutants (Fig. 6C). Retrograde transport in NT cells had an average velocity of 2.6 μm/s but was 6 times slower (0.32 μm/s) in V144D cells (Fig. 6D). Representative Supplementary Videos S1–S3 files (.avi) are provided for NT, WT, and V144D groups.
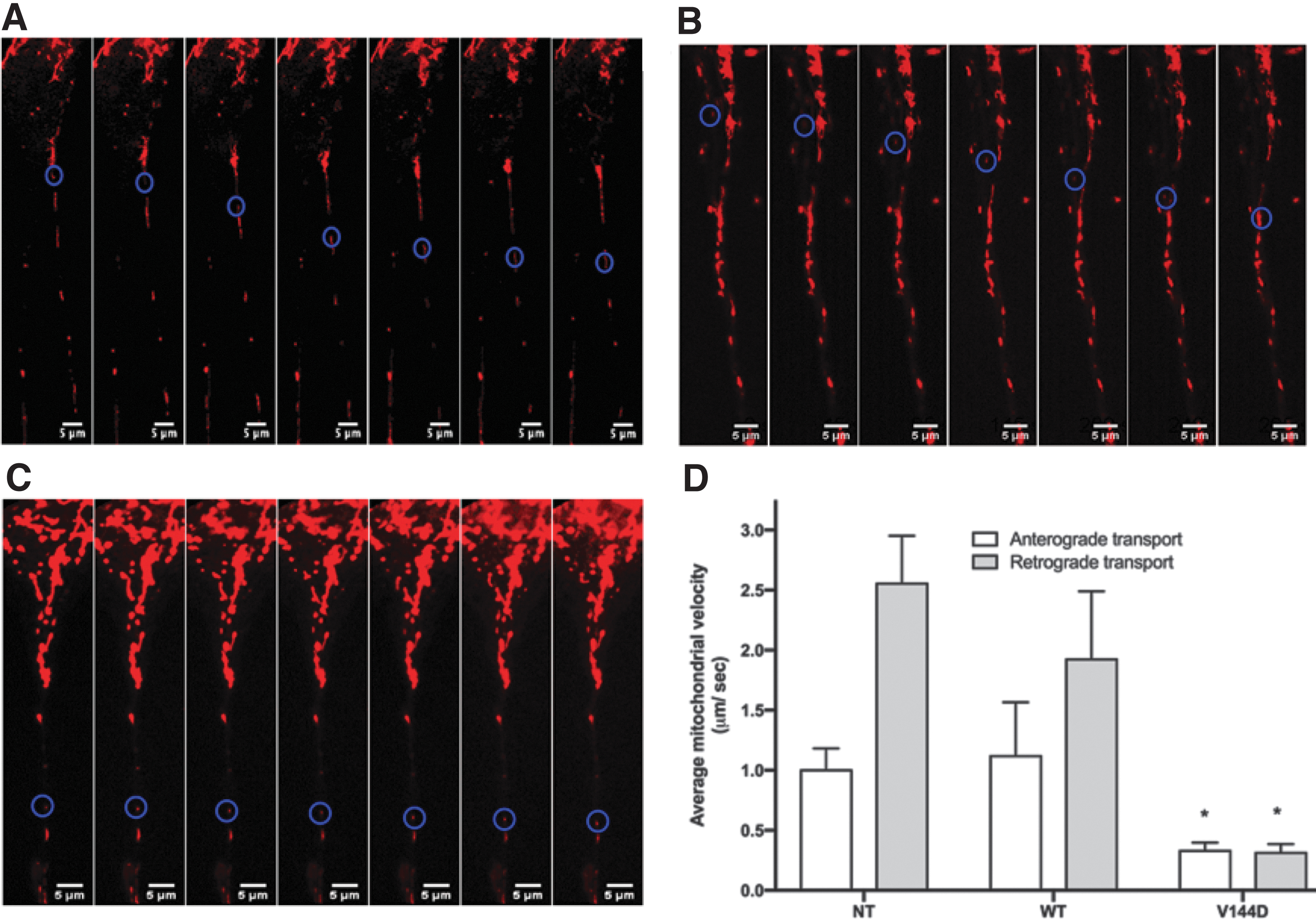
Measurement of average anterograde and retrograde mitochondrial velocity in neurite outgrowth of cells. Cells were treated with 50 nM Mitotracker-Red for 30 min and mitochondrial movements were measured by acquiring time-series images using a confocal microscope. The images were then analyzed to plot trajectories of individual mitochondria and calculate average velocity; maximal velocities in each group were recorded. Determination of mitochondrial velocity in both anterograde and retrograde directions were calculated using “Multiple Kymograph” plugin (EMBL Heidelberg), in Fiji (ImageJ). Mitochondria with velocities 0.1 μm/s or slower were considered sedentary and excluded from the measurements. Velocities were tracked in anterograde and retrograde directions and the data plotted. Representative time-series images of NT
Discussion
HSN1 is a length-dependent axonal neuropathy characterized by distal sensory and motor axonal degeneration, causing retrograde degeneration. Previous studies in our laboratory using ND15 cells (a DRG-derived cell line) showed increased cytoplasmic Ca2+ in HSN1 V144D mutants (Stimpson et al., 2016). Studies in V144D patient lymphoblasts have also shown remarkable mitochondrial structural aberrations and disrupted ER homeostasis, revealing links to Ca2+ dysregulation (Myers et al., 2014).
Furthermore, exogenously supplied deoxysphingoid bases caused rapid depletion of ER Ca2+ and loading of mitochondrial Ca2+ in primary cultures of motor and sensory neurons (Wilson et al., 2018). Nevertheless, a direct link between Ca2+ dysregulation and defective axonal transport has not yet been established in HSN1. Here we establish that the V144D mutation in SPTLC1 results in increased calpain and proteasome activities and disrupts the normal microtubule architecture, impairing the binding of the microtubule-associated proteins MAP2, tau, and motor proteins.
Ca2+ imbalance has long been implicated in the pathogenesis of neurodegenerative processes (Panov et al., 2002; Wallace, 2005; Elfawy and Das, 2019). Spatial and temporal plasticity of Ca2+ signaling makes it a versatile system, requiring intricate regulatory mechanisms; disruption can lead to the apoptotic or autophagic responses seen in many neurodegenerative disorders (Wallace, 2005; Hajnóczky et al., 2006). During cytosolic Ca2+ dysregulation, mitochondria play a significant role, acting as repositories for excess Ca2+, increasing uptake through the mitochondrial Ca2+ uniporter (MCU) (Berridge et al., 2003; Fifre et al., 2005).
Subsequently, mitochondrial Ca2+ homeostasis is restored by opening of the permeability transition pore (PTP) and the resulting discharge of Ca2+ leads to transient depolarization of the mitochondrial membrane potential (ΔΨm) (Kirichok et al., 2004; Woods et al., 2019). Thus, mitochondrial Ca2+ overload and depletion may be viewed as two sides of the same coin, and are highly temporal. Sustained opening of the PTP can also result in solute accumulation in mitochondria and the consequent expansion and rupture of the mitochondrial matrix (Hajnóczky et al., 2006; Starkov, 2010). In the current study, we observed a decrease in mitochondrial Ca2+ and an increase in cytosolic Ca2+ in V144D mutant cells. Emptying of mitochondrial Ca2+ into the cytosol has been shown to activate several Ca2+-dependent enzyme systems (Ruiz-Meana et al., 2006).
Calpain is a Ca2+-activated nonlysosomal protease, the controlled activation of which facilitates cytoskeletal remodeling, cell differentiation, cell migration, and an array of normal cell functions. However, pathological increases in cytoplasmic Ca2+ and calpain activation result in erratic proteolysis of cytoskeletal proteins causing disruption of cytoskeletal architecture (Khorchid and Ikura, 2002; Camins et al., 2006). Our data confirm a strong increase in calpain (I and II) and 20S proteasome activities, which are both independently triggered by transient increases in cytosolic Ca2+ (Park et al., 2013), in V144D cells.
Calpain activity was also confirmed by an increase in breakdown products of spectrin α II (SBP), a calpain substrate. Increased cytoplasmic Ca2+ and calpain activation was also detected in WT cells overexpressing SPTLC1 protein, although it was not accompanied by decreased mitochondrial Ca2+ or increased SBP accumulation. Calpain has several substrates in neuronal cells, among which MAP2 and tau function as important regulators of microtubule dynamics (Johnson et al., 1991; Ferreira and Bigio, 2011).
Microtubules are stabilized by the binding of MAPs, which prevent microtubule depolymerization, thus maintaining axonal structure and stability (Sánchez, 2000). We observed a significant decrease in cytosolic MAP2 levels, consistent with the increase in calpain activity, suggesting calpain-mediated degradation of MAP2 in V144D cells. We also confirmed calpain-mediated cleavage of MAP2, tau, and spectrin α II in the presence of Ca2+, which was reversed in the presence of calpain inhibitor, thus confirming the role of disruptive role of calpains in the presence of the V144D mutation.
Similarly, lower MAP2 and tau levels were also observed in the microtubule fractions from V144D mutants, providing strong evidence of impairment to microtubule ultrastructure; notably, these proteins play pivotal roles in microtubule dynamics, axonal transport, and neurite outgrowth (Zhang and Dong, 2012). Thus, the current findings implicate impaired MAP2 and tau availability as the most likely cause, resulting in severing along microtubule tracks, rendering them incapable of facilitating long distance transport.
Microtubule-binding capacity of both MAP2 and tau is also regulated by phosphorylation (Sánchez, 2000). Motor proteins were also found to be affected by the V144D mutation. Motor proteins facilitate the intricate bidirectional mechanism of axonal cargo transport (e.g., membranous organelles and large protein complexes) along the length of microtubules. Here, dynein, responsible for retrograde transport along the axons, was markedly increased in the microtubule fraction of the V144D mutants, suggesting inefficient retrograde transport.
Investigations conducted by Strom et al. (2008) indicate that disruption of the basal levels of dynein and/or dynactin severely compromise function and health of neurons including both long axons and motor neurons with one such function being the impairment of retrograde transport. It is thus likely the cells respond to decreased cargo transport by engaging more dyneins to the microtubules in an attempt to maintain retrograde transport.
In contrast, KIF2A, a microtubule depolymerizing motor protein, was found to be less engaged with microtubules from the V144D mutant relative to the controls. KIF2A is responsible for regulation of microtubule length and suppression of collateral axonal branch extension in neuronal cells (Homma et al., 2003); thus, a significant decrease in KIF2A would disrupt microtubule dynamics in the cells, although exact mechanisms are not known. Further to this, the data indicate a significant decrease in bidirectional axonal mitochondrial transport velocities in the V144D mutants.
Taken together, these findings suggest that Ca2+ dysregulation plays a significant role in disrupting the structure and hence functional capability of the microtubule cytoskeleton in V144D mutants by activating Ca2+-dependent proteases and subsequently cleaving crucial microtubule stabilizing proteins. The resulting decreased binding of microtubule-associated proteins then limits long distance cargo transport, and thus heavily contributes to progression of the pathology (Fig. 7).

Schematic representation of proposed molecular pathway in HSN1 characterized by axonal dying back. Calcium dysregulation and increased cytoplasmic Ca2+ as a result of the HSN1 mutation results in increase in calpain activity and subsequent breakdown of its substrates, the microtubule stabilizing proteins: MAP2 and tau. This potentially disrupts the axonal microtubule track, derailing the pathways of motor proteins such as kinesins and dynein, disturbing long-distance transport. Consequent decrease in transportation of mitochondria, other membrane-bound organelles, lipids, and synaptic vesicles to and from the cell body, may lead to axonal dying back and neurodegeneration. Color images are available online.
Pathologies such as diabetic neuropathy (Kharatmal et al., 2015) and taxol-induced neuropathy (Wang, 2003) have symptomology similar to HSN1, and calpain has been shown to play a significant role in their molecular underpinnings; however, the exact role of calpain in disease progression has never been previously demonstrated.
Calpain inhibitors MDL28170 and AK295 have been found to significantly improve nociceptive and biochemical deficits in diabetic sensory neuropathy (Kharatmal et al., 2015) and taxol-induced sensory neuropathy (Wang, 2003), suggesting calpain as a likely therapeutic target. Consistent with this, our data provide a better understanding of the molecular mechanism leading to axonal degeneration in HSN1 and possibly other neurodegenerative disorders by establishing a link between cytosolic and mitochondrial Ca2+ dyshomeostasis, calpain activation, disruption of microtubule structure, and long-distance axonal transport, bringing us one step closer to finding optimal targets for potential new therapeutics that could slow down or even reverse this neurodegenerative pathway.
Conclusion
We have established a link between cytosolic and mitochondrial Ca2+ dyshomeostasis, calpain activation, disruption of microtubule structure and long-distance transport of mitochondria in neurites. Further studies would help in identifying potential drug targets that could slow down or even reverse this neurodegenerative pathway.
Footnotes
Acknowledgment
The authors thank Dr Sindy Kueh for the technical support in confocal microscopy.
Authors' Contribution
S.M., A.A. and J.R.C. conceived the initial study and designed the experiments. A.A. performed the experiments and S.M. supervised the project. N.N. assisted with the calcium studies. A.A. and A.L. performed statistical analysis on the data. A.A. wrote the article with input from S.M. and J.R.C. All authors read and edited the article.
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure Statement
The authors declare no competing financial interest.
Funding Information
A.A. acknowledge support from the Researcher Development Strategic Initiative Funding—New Staff Scheme, Western Sydney University. S.M. acknowledges the support of a private anonymous philanthropic foundation.
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.