
Abstract
Apoptosis, necroptosis, and autophagy are the major programmed cell death in myocardial ischemia–reperfusion injury (MIRI). Maslinic acid (MA) has been found to regulate pathophysiological processes that mediate programmed cell death in MIRI, such as inflammation and oxidative stress. However, its effects on MIRI remain unclear. This study intends to explore the role of MA in MIRI. In vitro, MA had no obvious cytotoxic effects on H9C2 cells, and significantly improved the impaired cell viability caused by hypoxia reoxygenation (HR). In vivo, MA significantly alleviated ischemia reperfusion (IR)-induced left ventricular myocardial tissue injury, downregulated creatine kinase-myocardial band (CK-MB), and lactate dehydrogenase (LDH) levels in serum as well as reducing infarct size. Moreover, MA inhibited HR-induced mitochondrial apoptosis and necroptosis in vitro and in vivo. Of interest, MA interacts with lysosome-associated membrane protein 2 (LAMP2). MA protected LAMP2 from IR and promoting autophagic flux to inhibit apoptosis and necroptosis, whereas these effects were reversed by co-treatment with lysosomal inhibitor BarfA1. In conclusion, MA can inhibit MIRI-induced apoptosis and necroptosis by promoting autophagic flux. These results support that MA is a potential agent to ameliorate MIRI.
Introduction
Acute myocardial infarction is the main cause of death of cardiovascular diseases (Anderson, 2017). Reperfusion therapy can quickly restore the blood supply for ischemic myocardium to reduce the mortality during hospitalization, but the subsequent myocardial ischemia–reperfusion injury (MIRI) will cause further myocardial injury that leads to ∼50% final infarct size (Mcalindon et al., 2015; Heusch, 2020). However, there is currently no effective treatment strategy for MIRI. Exploring the mechanism of cardiomyocytes death in MIRI will contribute to exploring safe and effective agents for treatment.
The primary harm of MIRI causes substantial cardiomyocytes death, which impairs the pump ability of heart (Del Re et al., 2019). Cell necrosis is the main mode of death during the ischemic period, and autophagy, apoptosis, and necroptosis are major programmed cell death during reperfusion period (Linkermann et al., 2013; Teringova and Tousek, 2017; Zhu et al., 2018). Experimentally, Koshinuma et al. (2014) demonstrated that simultaneous inhibition of necroptosis and apoptosis by their specific inhibitors (Necrostatin-1 and Z-VAD) can further enhance the cardioprotective effect compared with inhibiting necroptosis or apoptosis alone. However, previous studies have hardly found a key mechanism that can simultaneously regulate apoptosis and necroptosis (Lee et al., 2012; Hu et al., 2020). Therefore, it is necessary to explore this regulatory mechanism and its treatment drug.
Autophagy has been found to have a double-edged sword effect with promoting “survival” and “death” in MIRI (Matsui et al., 2007; Nah et al., 2016). During ischemia period, cardiomyocytes activate autophagy upon lack of nutrient metabolites, and then degrade unnecessary proteins and organelles to produce amino acids and fatty acids to maintain energy supply, thereby reducing cell necrosis (Shi et al., 2019). However, during reperfusion, cardiomyocytes are affected by oxidative stress, inflammatory, and organelle dysfunction, hyperactivating Beclin-1-mediated autophagic death (Matsui et al., 2008). Of interest, in this process, autophagy-related proteins can act as scaffold for apoptosis and necroptosis. Beclin-1 is a key protein for mediating the production of autophagosomes, and has been proven to bind to Bcl-2 through the homologous BH domain to promote apoptosis (Dong et al., 2019).
Moreover, P62 mainly mediates the fusion of autophagosomes and lysosomes to degrade the substances phagocytosed and has been proved that it can also mediate the phosphorylation of RIPK1 and RIPK3 to activating necroptosis (Liu et al., 2018a). Some studies have shown that impaired autophagic flux in MIRI causes autophagy to shift from mediating survival to promoting death (Ma et al., 2012a, 2012b; Wang et al., 2019). Impaired lysosomal structure and effects is the key factor of downregulated autophagic flux in MIRI, and remote ischemic preconditioning and gene therapy can promote autophagic flux by protecting lysosomes to reduce the autophagic death (Fan et al., 2014; Song et al., 2019). These studies indicate that effectively protected lysosomes may promote autophagic flux, reduce autophagic death, even inhibiting apoptosis and necroptosis at the same time.
Maslinic acid (MA) is a compound of pentacyclic triterpene, first extracted from olive pomace, and naturally present in skins of many fruits and vegetables such as hawthorn and olive (Nagai et al., 2019; Lee et al., 2020). Studies have reported that MA can suppress inflammation and redox stress, and alleviate many types of inflammatory disease such as acute lung injury (Jeong et al., 2020) and arthritic diseases (Fukumitsu et al., 2016). Many studies have demonstrated that MA can protect cardiomyocytes from hyperglycemia (Hung et al., 2015), pressure overload (Liu et al., 2018b), and isoproterenol insults (Hussain Shaik et al., 2012), implying MA possesses great cardioprotective effects. Of interest, MA has the ability to regulate autophagy and inhibit apoptosis (Dong et al., 2017). Moreover, we found that MA has a direct binging ability with the lysosome-associated membrane protein 2 (LAMP2) based on pharmacological analysis. Therefore, we hypothesized that MA may inhibit apoptosis and necroptosis by affecting autophagic flux in MIRI.
Materials and Methods
Cell culture and hypoxia reoxygenation model
H9C2 cell line was purchased from National Collection of Authenticated Cell cultures (Serial: GNR 5). Cells were cultured with complete medium containing 90% Dulbecco's modified Eagle's medium (DMEM; Gibco) and 10% fetal bovine serum (Bioind) at 37°C and 5% CO2 incubator. Cells were seeded in cell plate or dish when the cell density reaches 80% for 1 day before any further treatment; 10 mM × 5 mL MA was purchased from the MedChemExpress (Serial: HY-N0629), and diluted with complete medium to 0, 1, 5, 10, 20, 40 μM. H9C2 cells were pretreated with MA for 24 h before hypoxia reoxygenation (HR) treatment. The AnaeroPack system (Mitsubishi Gas Chemical Company) was used to construct an oxygen-deficient atmosphere with 95% N2 and 5% CO2 as described previously (Liu et al., 2021). In brief, H9C2 cells were cultured in serum-free DMEM, and placed in the AnaeroPack incubator for 4 h at 37°C, and then moved to a 95% O2 and 5% CO2 incubator for 4 h at 37°C. The control group was cultured at 95% O2 and 5% CO2 incubator for 8 h.
Rats MIRI model establishment
Sixty adults-specific pathogen-free male Sprague-Dawley rats, weighing 250–300 g, were purchased from China Three Gorges University (Yichang, China). The animals were acclimatized under controlled conditions of humidity (50% ± 10%), room temperature (22°C ± 1°C), and light/dark cycle (12 h/12 h) in Animal Center of China Three Gorges University. All the rats had free water and food during the experimental period. The protocols of this study were approved by China Three Gorges University [Approval No. SYXK (HUBEI) 2017-0061]. The rats were randomly divided into Sham+dimethyl sulfoxide (DMSO) group (n = 12), Sham+MA group (n = 12), IR+DMSO group (n = 12), IR+MA group (n = 12), and IR+MA+BafA1 group (n = 12) by random number table method. Forty-eight hours before myocardial ischemia–reperfusion operation, rats were injected intraperitoneally with 20 mg/kg of MA or 1 mg/kg of bafilomycin A1 (BafA1) or the same amount of DMSO dissolved in normal saline according to the grouping, and the injection was repeated 24 h before surgery. Rats were anesthetized by 3% sodium pentobarbital, and its breathing was maintained by small animal ventilator. The left anterior descending (LAD) coronary artery was ligated by 6-0 silk with 1 mm medical latex tube for 30 min to induced myocardial ischemia; then the suture was cut to restore blood flow of LAD for 3 h. The Sham group was subjected to the same operation without LAD occlusion. Blood samples were collected for measuring lactate dehydrogenase (LDH) and creatine kinase-myocardial band (CK-MB) by automatic biochemical analyzer (Siemens, Berlin, Germany).
Cell proliferation assay
Both drug toxicity and cell viability were assayed by Cell Counting Kit-8 (CCK-8) kit (Serial: CK04; Dojindo). In brief, cells were seeded in 96-well plate with 1 × 104 cells/well. After cells were treated with 0, 1, 5, 10, 20, and 40 μM of MA for 24 h, 10 μL CCK-8 was added in each well and incubated for 2 h to examine the drug toxicity. After HR treatment, 10 μL CCK-8 was added in each well and incubated for 2 h to examine the cell viability. The absorbance values were measured by microplate reader at 450 nm.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was purchased from Beyotime (Serial: C1086 for cells, C1089 for myocardial tissue). In vitro, cells were seeded in the laser confocal cell culture dishes with 1 × 105 cells/dish. After HR treatment, cells were fixed with 4% formaldehyde for 30 min, treated with immunostaining permeabilization solution for 10 min, and then stained with TUNEL working solution for 1 h, and stained with DAPI for 10 min. In vivo, after IR model establishment, myocardial tissues were prepared as paraffin sections. Each section was blocked for 2 h and permeabilized for 15 min, then incubated with α-actin (Serial: AF0048; Beyotime) overnight. After washing off the primary antibody, the section was stained by Alexa Fluor 488 and TUNEL solution at 37°C for 1 h, and stained with DAPI for 10 min. The fluorescents were observed by laser confocal microscope (Leica DMi8).
Necroptosis analyses
In vitro, DAPI and PI solution were used to detect cell necroptosis; these reagents were purchased from Beyotime (Serial: C1005 and ST511). Cells were seeded in six-well plate with 1 × 106 cells/well, and directly stained by DAPI/PI solution for 10 min after HR treatment. The blue fluorescence and red fluorescence were observed under fluorescence microscope.
In vivo, p-MLKL (No. 91689; Cell Signaling Technology) immunohistochemistry staining was conducted to detect myocardial necroptosis. Paraffin sections of myocardial tissue were blocked by 10% bovine serum albumin, incubated with the primary antibody at 4°C overnight, incubated with the HRP-labeled secondary antibody at 37°C for 1 h, and finally observed by microscope.
Hematoxylin and eosin staining
Myocardial tissue was embedded in paraffin, cut into 3–5 μm sections, and stained by hematoxylin and eosin (H&E) staining solution (C0105S; Beyotime Company). Then the sections were observed under a light microscope.
Molecular docking simulation
The AutoDock software was used to dock MA and LAMP2. The chemical structure of MA was downloaded from PubChem (CID: 73659), and molecular structure of LAMP2 was downloaded from RCSB (5GV3). Before docking, water molecules were deleted, and polar hydrogen atoms were added. After docking by AutoDock, result was visualized by PyMOL.
Western blotting
The total protein from cells were lysed by RIPA buffer with 1% NaF, 1% Na3VO3, and 1% PMSF (G2002, G2007, and G2008; Servicebio). Ten percent SDS-PAGE gel was used to separate the proteins, and migrated to PVDF membrane electrophoretically. Five percent slimmed milk powder was dissolved in TBST solution, and used to block the membranes for 2 h.
After that, the membranes were incubated with primary antibodies overnight at 4°C, including Bax (50599-2-Ig; Proteintech), Bcl-2 (12789-1-AP; Proteintech), cleaved Caspase-9 (No. 20750; Cell Signaling Technology), cleaved Caspase-3 (No. 9661; Cell Signaling Technology), p-RIPK1 (No. 65746; Cell Signaling Technology), p-RIPK3 (No. 93654; Cell Signaling Technology), p-MLKL (No. 91689; Cell Signaling Technology), LAMP2 (No. 49067; Cell Signaling Technology), Beclin-1 (No. 3495; Cell Signaling Technology), P62 (No. 23214; Cell Signaling Technology), LC3I/II (No. 12741; Cell Signaling Technology), and β-actin ( GB11001; Servicebio). Then the membranes were incubated with secondary antibodies (GB23301; Servicebio) for 2 h at room temperature. Finally, the membranes were visualized by the super sensitive ECL luminescent liquid (Series: P1050; Applygen).
Statistical analysis
The software SPSS 20.0 was used for data analysis. All values were expressed as mean ± standard deviation (SD). One-way analysis of variance analysis followed by least significant difference post hoc test was used for comparisons between groups. A value of p < 0.05 was regarded as the difference is statistically significant.
Results
Effect of MA on HR-induced cardiomyocyte injury
To determine the possible drug toxicity of MA on H9C2 cells, H9C2 cells were treated with different concentrations of MA from 1 to 40 μM for 24 h, and then CCK-8 kit was used to detect the cell viability. As given in Figure 1B and C, MA has no significant drug toxicity for H9C2 for 24 h, and even slightly increased cell viability, although the difference between the groups was not statistically significant. To mimic model of MIRI in vitro, we established a model of hypoxia for 4 h followed by reoxygenation for 4 h. The data showed that HR significantly impaired cell viability compared with the control group, and MA markedly attenuated HR-induced cell injury when concentration was from 5 to 40 μM, which showed a certain dose dependency. Compared with 20 μM of MA treatment, 40 μM of MA could not provide further cardioprotection (p > 0.05). These results suggested that MA has no obvious cytotoxic effect on H9C2 cells, and can significantly improve the impaired cell viability caused by HR.
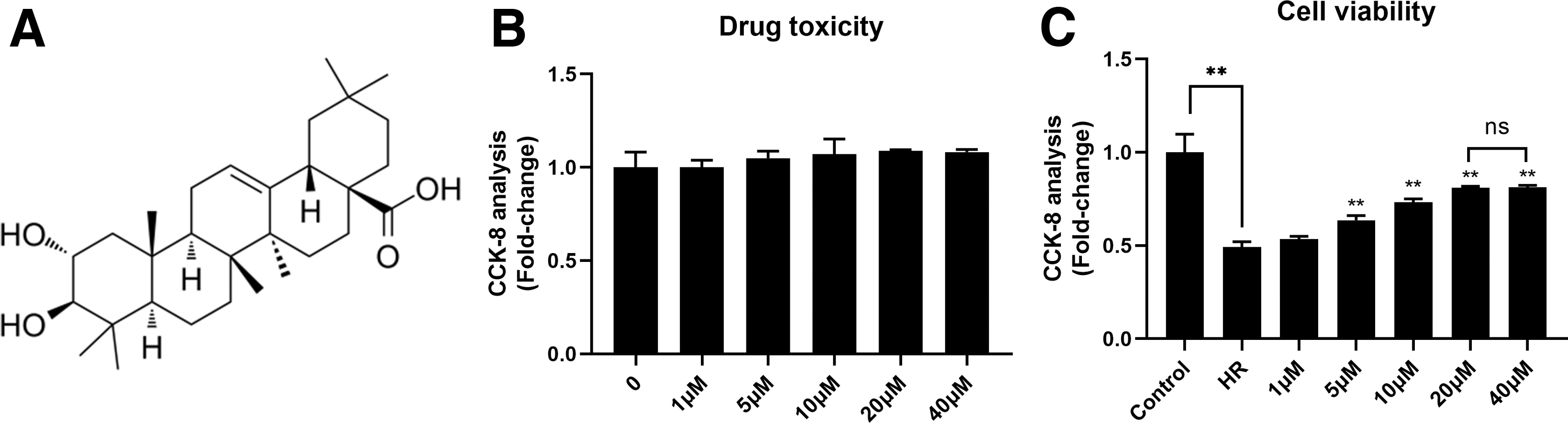
MA can alleviate HR-induced cell injury.
Effect of MA on HR-induced cell apoptosis in vitro
To determine the myocardial protective effect of MA, we first detected the level of cell apoptosis in each group by TUNEL staining. As given in Figure 2A, the results of laser confocal microscopy showed that very few TUNEL-positive cells existed in the control group, and a large number of TUNEL-positive cells were detected after HR insult. However, treatment with MA could significantly reduce the number of TUNEL-positive cells, implying that MA can downregulate the level of apoptosis induced by HR.
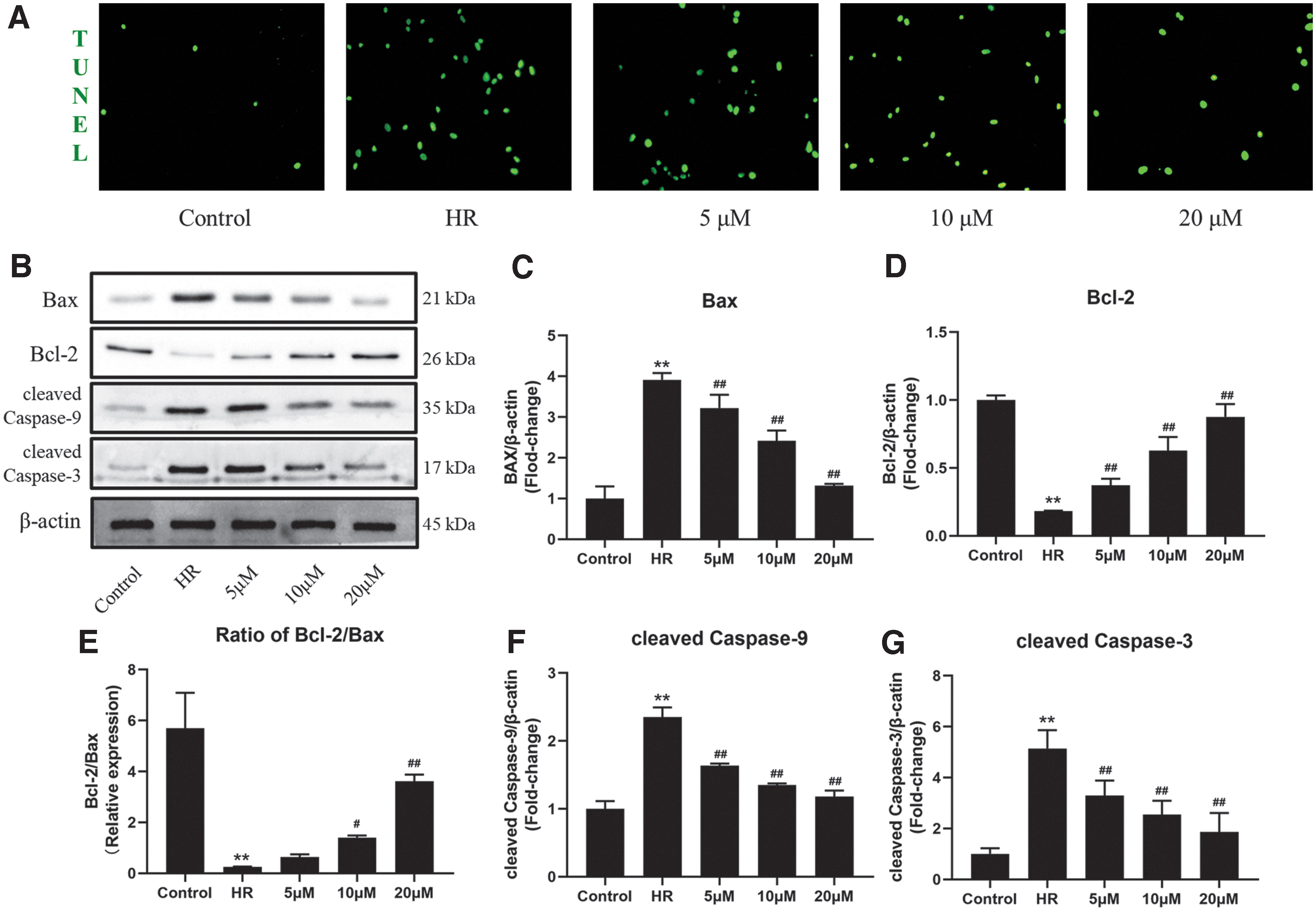
MA can significantly inhibit HR-induced mitochondrial apoptosis in vitro.
To further investigate the anti-apoptotic mechanism of MA, we tested the expression of endogenous apoptosis-related proteins by Western blotting (Fig. 2B–G). Compared with the control group, expressions of Bax and level of cleaved Caspase-3 and cleaved Caspase-9 were markedly upregulated, whereas Bcl-2 expression was significantly decreased in the HR group. Compared with the HR group, the expressions of Bax and level of cleaved Caspase-3 and cleaved Caspase-9 were significantly decreased by MA in a dose-dependent manner. These results suggested that MA can inhibit HR-induced mitochondrial apoptosis.
Effect of MA on HR-induced necroptosis in vitro
To detect the effect of MA on cell necroptosis, we detected the ratio of necrosis by the DAPI/PI staining. As given in Figure 3A, HR induced a great number of PI-positive cells. However, the groups treated with MA showed a gradual decrease in the PI-positive rate as the drug dose increased. Necrosis has always been regarded as a passive type of cell death that cannot be regulated, whereas necroptosis has recently been found to be a regulated type of necrotic cell death. Therefore, the result implies that MA may alleviate cardiomyocytes HR injury by inhibiting necroptosis.

MA can significantly inhibit HR-induced necroptosis in vitro.
The activation of RIPK1/RIPK3/MLKL mediates necroptosis; thus, we detected the phosphorylated form of these molecules. As given in Figure 3B–E, HR significantly increased the phosphorylation of RIPK1, RIPK3, and MLKL compared with the control group. MA-treated H9C2 cells showed decrease in levels of p-RIPK1, p-RIPK3, and p-MLKL after HR, compared with the HR group. It was suggested that MA alleviated cardiomyocytes HR injury partially by inhibiting necroptosis.
Effects of MA on IR-induced myocardial injury in vivo
To determine whether MA can alleviate the myocardial injury upon IR insulted, rats were subjected to IR or sham surgery with or without pretreating with MA. As given in Figure 4A and B, myocardial infarct size was stained by TTC solution or H&E staining, respectively. The results showed that I/R caused ∼30% infarct size, but MA treatment significantly reduced I/R-caused infarct size to 15% (Fig. 4C).
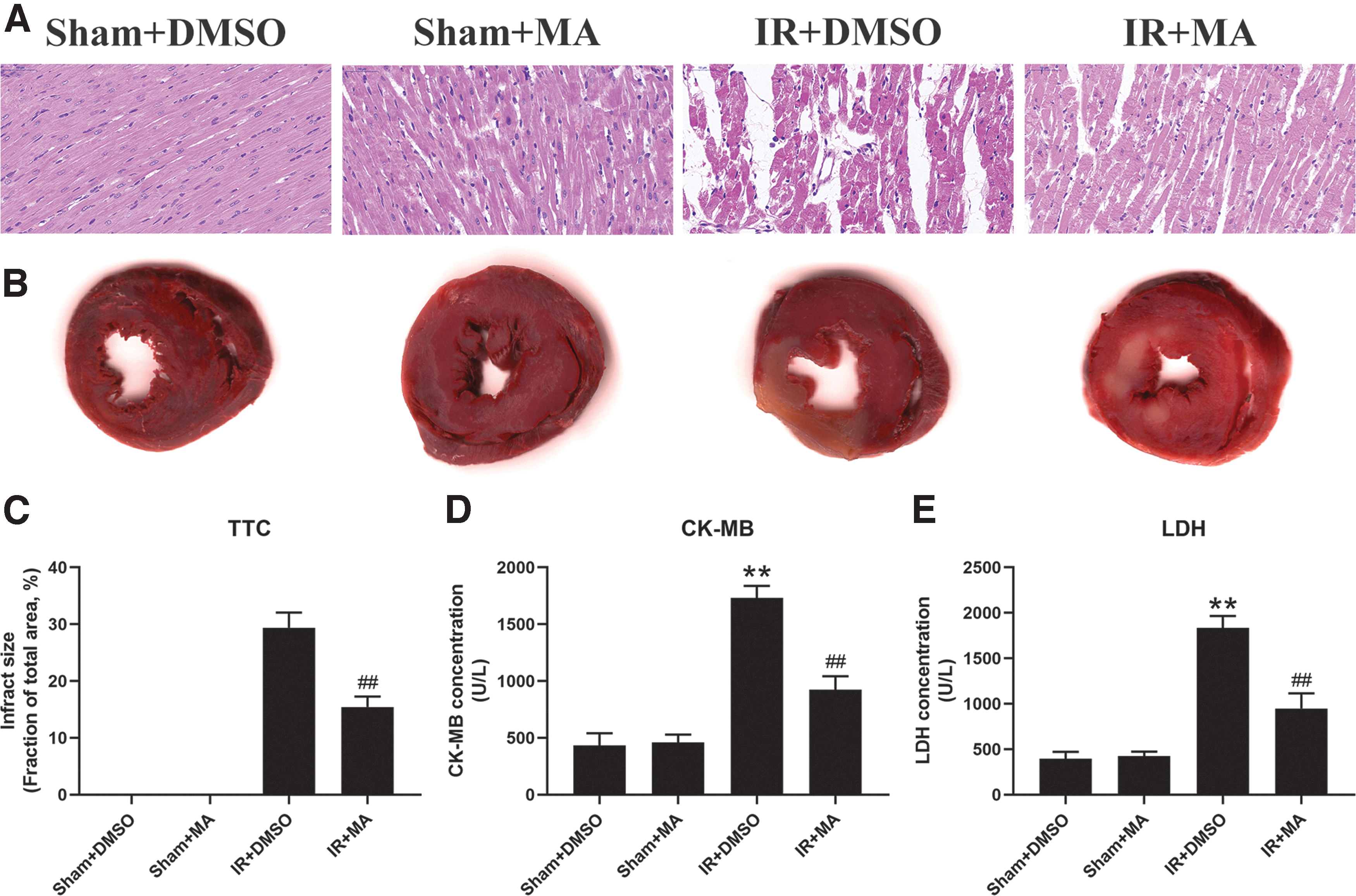
MA can significantly alleviate IR-induced myocardial injury in vivo.
Myocardial fiber breakage and cellular edemas as well as neutrophil infiltration were clearly observed in the IR group, but these changes were significantly alleviated in the MA+IR group. Furthermore, IR significantly increased the level of CK-MB and LDH compared with the sham group. However, these changes were significant reduced in MA-treated IR rats (Fig. 4D, E). Collectively, these data suggested that MA could alleviate IR-induced myocardial injury in vivo.
MA can inhibit IR-induced apoptosis and necroptosis simultaneously in vivo
Further exploring the mechanisms contributing to the cardioprotection of MA, we did TUNEL/α-actin double staining, MLKL immunohistochemistry staining, as well as measured protein markers associated with apoptosis and necroptosis.
As shown in Figure 5, the Sham+DMSO group and the Sham+MA group had few TUNEL-positive and MLKL-positive cells. TUNEL-positive staining in the IR+DMSO group not only appeared in cardiomyocytes, but also more in between cardiomyocytes, proving that IR-induced apoptosis occurred in cardiomyocytes, endothelial cells, and fibroblasts.
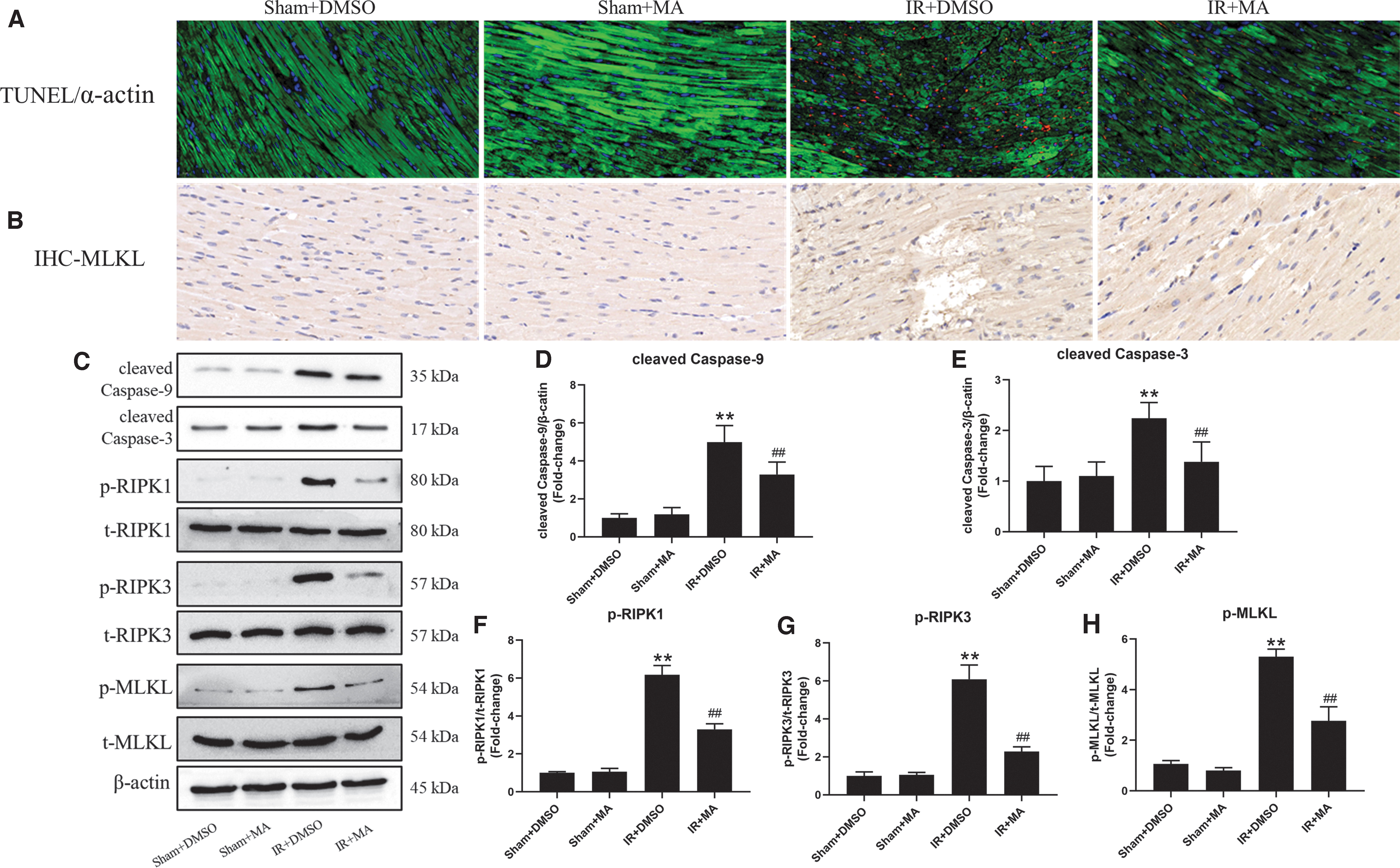
MA can inhibit IR-induced apoptosis and necroptosis simultaneously in vivo.
Of interest, rats pretreated with MA significantly reduced TUNEL-positive cells, and this protective effect did not show obvious cardiomyocytes specificity, suggesting that the anti-apoptotic effect of MA may be simultaneously acting on multiple cells present in the myocardial tissue. Consistent with the above results, IR significantly increased the expression levels of cleaved caspase-3, cleaved caspase-9, p-RIPK1, p-RIPK3, and p-MLKL, whereas pretreating with MA significantly inhibited the activation of these molecules induced by IR. These results suggest that MA can inhibit IR-induced apoptosis and necroptosis simultaneously.
MA can promote autophagic flux by docking LAMP2 while reversed by BafA1
We used the AutoDock software to examine the affinity of molecular docking. As given in Figure 6A, the binding energy value of LAMP2 and MA was −5.98 kcal/mol, suggesting that the binding is significant and stable. To further investigate the effect of MA on autophagic flux and lysosomal function, we measured molecules associated with autophagy and lysosome, and used the later stage lysosomal inhibitor BafA1, which principally block the fusion and degradation in lysosomes to block autophagic flux (Zhu et al., 2017).
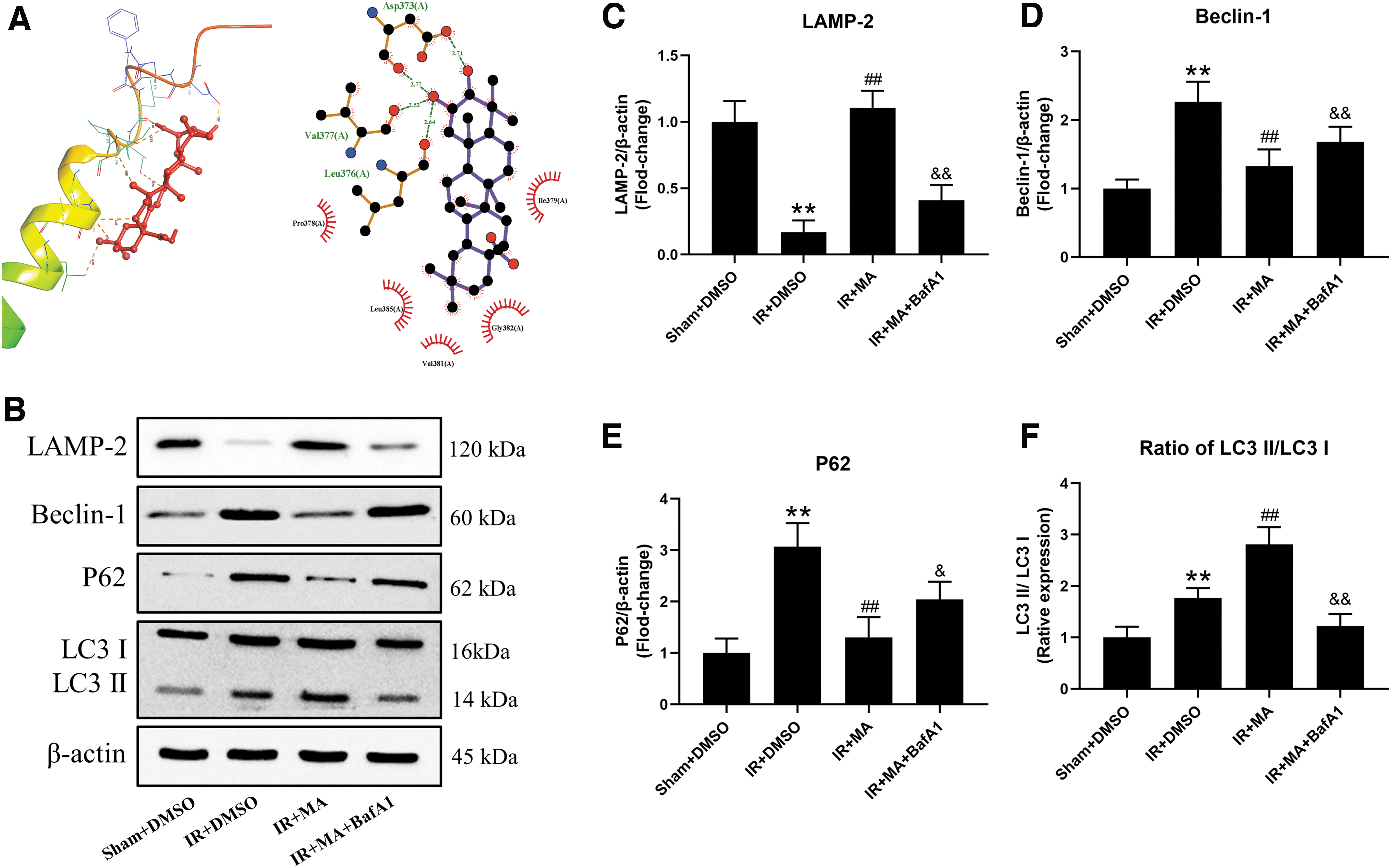
MA can promote autophagic flux by binding to LAMP2.
These western blotting results showed that IR significantly induced overexpression of Beclin-1, P62, and LC3II while downregulating LAMP2 expression, compared with the Sham+DMSO group. Of interest, pretreating with MA further increased expression of LC3II and LAMP2, whereas it reduced expression of Beclin-1 and P62. However, co-treating BafA1 and MA before IR lead to marked reduction in expression of LAMP2 and LC3II while upregulated expression of Beclin-1 and P62, compared with the IR+MA group. These data may demonstrate that IR impairs autophagic flux and lysosomal membrane structure. Treatment of MA could protect lysosome from MIRI and further promote autophagy flux.
MA can inhibit apoptosis and necroptosis while abolished by BafA1
Next, we measured expression of apoptosis and necroptosis molecules in myocardial tissue of each group. As given in Figure 7, MA can significantly downregulate levels of cleaved Caspase-3, cleaved Caspase-9, p-RIPK1, p-RIPK3, and p-MLKL induced by IR, but the protective effects of MA were partially abolished by BafA1. The above results suggest that IR induced excessive apoptosis and necroptosis simultaneously, but MA can effectively inhibit IR-induced apoptosis and necroptosis by promoting autophagic flux.

MA simultaneously inhibited apoptosis and necroptosis depend on autophagic flux.
Discussion
Many studies have identified a lot of treatment in experimental HR or IR models, but few of these studies translate into the clinical scenario. The main reason may be incomplete understanding of mechanisms underlying MIRI, which is multifactorial and cause many kinds of cell death (Hausenloy et al., 2017). Emerging consensus suggested that optimal cardioprotection may require the combination of synergistic multitarget (Davidson et al., 2019). In this study, we found that MIRI could cause excessive apoptosis and necroptosis both in vivo and in vitro, whereas pretreating with MA could effectively inhibit apoptosis and necroptosis. The protective mechanism of MA is associated with promoting autophagic flux, which could be blocked by lysosomal inhibitor BafA1, suggesting MA inhibits apoptosis and necroptosis depending on promoting autophagy and protecting lysosome.
MIRI involves a variety of physiological and pathological changes, including redox stress, calcium overloading (Chang et al., 2019), metabolic disorder, and finally activating programmed cell death (Heusch and Gersh, 2017). MA is the main pentacyclic triterpenes present in olives and olive oil, and has been reported by several experimental studies to have anti-inflammatory (Pavel et al., 2019) and antioxidant stress (Rufino-Palomares et al., 2016). Whether MA is an effective agent for alleviating MIRI remains unclear, which may limit further clinical use of MA.
This study revealed that MA can effectively alleviate MIRI and reduce the final infarct area. The myocardial protection mechanism of MA is associated with simultaneously inhibition of MIRI-induced apoptosis and necroptosis by promoting autophagic flux. Moreover, the safety of MA has been widely evaluated by acute and repeated oral administration in mice. The results showed that neither the toxicity nor side effects of MA as compared with the untreated group indicate that MA can be considered a safe drug (Sánchez-González et al., 2013). Our results also showed that MA has no obvious toxic effects on H9C2 cells and Sprague-Dawley rats. Therefore, MA is an effective and safe drug for the treatment of MIRI; these results filled the gap in the effects of MA on MIRI.
Apoptosis is the major type of programmed cell death in MIRI, and large number of studies have proven that inhibition of apoptosis by drug intervention or genetic technology can alleviate MIRI and decrease final infarct size (Li et al., 2020). Mitochondria are widely present in cardiomyocytes and are responsible for more than 90% ATP synthesis and the production of oxidation products. Mitochondria-mediated apoptosis is activated by overexpressed Bax, thereby promoting the Caspase-9/Caspase-3 cascade (Wang and Zhou, 2020). Bcl-2 protein has been identified as a key factor of anti-apoptosis in MIRI, as it can directly suppress the effect of Bax through their homologous BH-3 domain (Brocheriou et al., 2000).
Previous study demonstrated that MA can reduce the ratio of Bax/Bcl-2 to inhibit apoptosis induced by β-amyloid peptide in PC12 nerve cell line (Yang et al., 2015). In this study, HR significantly increased the expression of Bax, whereas it decreased the expression of Bcl-2, thereby promoting the cleavage of Caspase-9 and Caspase-3 to activate apoptosis. The result of TUNEL staining suggested that MA has a potent inhibitory effect on apoptosis. In line with the change, the results of western blotting showed that MA effectively downregulated expression of Bax, cleaved Caspase-9, and cleaved Caspase-3, and upregulated expression of Bcl-2 in vitro. The same anti-apoptotic effect of MA was overserved in rat IR model. Therefore, MA can alleviate MIRI partially by inhibiting apoptosis.
Necroptosis has been confirmed as another major type of programmed cell death in MIRI (Zhu and Sun, 2018). Necroptosis is initiated by phosphorylation of RIPK1, which is a protein kinase that can further phosphorylate RIPK3. Activation of RIPK3 can promote mitochondrial member transition pore open to expand the signal of necroptosis, and also phosphorylate MLKL to translocate to the cellular inner membrane for causing membrane rupture (Pouwels et al., 2016; Chen et al., 2020). Some studies have shown that apoptosis and necroptosis always occur in MIRI at the same time, suggesting that inhibition of apoptosis and necroptosis alone may not be enough to alleviate MIRI in clinical scenario (Koshinuma et al., 2014; Zhang et al., 2016; Zhu et al., 2018).
Therefore, the development of drugs that can simultaneously inhibit apoptosis and necroptosis has more clinical application value. Unlike apoptosis and autophagy, the integrity of the cell membrane of necrosis and necroptosis is destroyed, which allow the nuclear dye PI to enter the cells. In this study, the DAPI/PI double staining confirmed that MA can attenuate HR-induced necrotic death, and the mechanism is associated with decreased activity of RIPK1/RIPK3/MLKL axis. Moreover, the result of MLKL immunohistochemistry staining also showed MA could inhibit IR-induced necroptosis and reduced activity of RIPK1/RIPK3/MLKL axis. To our knowledge, our study is the first to identify the effect of MA on necroptosis.
Autophagy disorders can be observed in almost all cardiovascular disease, including but not limited to MIRI, hypertrophic cardiomyopathy, and heart failure (Mialet-Perez and Vindis, 2017). Until now, it is still a matter whether accumulation of autophagosomes in cardiomyocytes in MIRI means upregulation of autophagy or a consequence of blockade of autophagic flux (Gottlieb and Mentzer, 2013). Matsui et al. (2007) demonstrated that inhibition of autophagy exacerbated cardiomyocyte death in ischemia period, but relieved cell injury in reperfusion period. Ma et al. (2012b) suggested that MIRI downregulated LAMP2 transcript and expression thereby impairing autophagic flux, resulting in secondary cell programmed death to remove dysfunctional cardiomyocytes.
In this study, we found that IR downregulated LAMP2 expression, but upregulated LC3II and P62, indicating that formation of autophagosomes is promoted but the autophagic flux was blocked. The result of molecular docking showed MA can stably bind to LAMP2. Moreover, results of western blotting also showed pretreating with MA could protect LAMP2 from IR and downregulated P62 expression, whereas these effects were reversed by lysosomal inhibitor BarfA1, indicating MA has the ability for promoting autophagosome–lysosome fusion and degradation to improve autophagic flux. Moreover, MA inhibited IR-induced apoptosis and necroptosis simultaneously, but these effects were also abolished by co-treating with BafA1. These results suggested that MA could protect lysosome function to promote autophagic flux while inhibiting apoptosis and necroptosis simultaneously.
In conclusion, our results demonstrated that MA could effectively reduce cardiomyocytes death induced by IR. MA can inhibit MIRI-induced apoptosis and necroptosis by promoting autophagic flux. These results support that MA is a potential agent to ameliorate MIRI.
Footnotes
Disclosure Statement
Authors declare no conflict of interest.
Funding Information
This study was founded by the Fundamental Research Funds for the Central Universities (Grant No. 2042020kf0100 to S.S.).