
Abstract
Inflammation is a double-edged sword. The moderate inflammatory response is a fundamental defense mechanism produced by the body's resistance to dangerous stimuli and a repair process of the body itself. Increasing studies have confirmed that the overactivation of the inflammasome is involved in the occurrence and development of inflammatory diseases. Strictly controlling the overactivation of the inflammasome and preventing excessive inflammatory response have always been the research focus on inflammatory diseases. However, the endogenous regulatory mechanism of inflammasome is not completely clear. The tripartite motif (TRIM) protein is one of the members of E3 ligases in the process of ubiquitination. The universality and importance of the functions of TRIM members are recognized, including the regulation of inflammatory response. This article will focus on research on the relationship between TRIMs and NLRP3 Inflammasome, which may help us make some references for future related research and the discovery of treatment methods.
General Introduction
The inflammasome is an important part of the innate immune system. Martinon's research group first proposed the concept of the inflammasome in 2002. It refers to a polyprotein complex assembled by cytoplasmic pattern recognition receptors (PRRs), also known as receptor proteins, adaptor protein ASC (apoptosis-associated speck-like protein containing CARD), and Pro-Caspase-1 (Martinon et al., 2002). Assembly of the inflammasome is the body's stress response to pathogen-associated molecular patterns (PAMPs) and dangerous-associated molecular patterns (DAMPs). The successfully assembled inflammasome can activate Caspase-1 to become Cleaved Caspase-1 with shear function.
On the one hand, Cleaved Caspase-1 can shear Pro-IL-1β and Pro-IL-18 to become IL-1β and IL-18 mature (Cerretti et al., 1992; Thornberry et al., 1992; Ghayur et al., 1997; Gu, 1997). On the other hand, cleaved Caspase-1 cleaves GSDMD, a member of the Gasdermin family, to form a peptide containing Gasdermin-N-terminal active domain. The peptide of the Gasdermin-N-terminal active domain can punch holes in the cell membrane composed of natural phospholipids, and form many honeycomb channels with a diameter of about 14 nm.
Large amounts of IL-1β, IL-18, and cell contents can be released into the extracellular environment from these holes, recruiting inflammatory cells to gather, causing an inflammatory response (Ding et al., 2016; Shi et al., 2017). At the same time, cells continue to swell to rupture, leading to cell death. This way of cell death, which will be regulated by forming plasma membrane pores relying on Gasdermin family proteins under the pathological conditions of inflammation and stress, is called pyroptosis (Boise and Collins, 2001; Galluzzi et al., 2018). Therefore, cleaved Caspase-1 is also known as the effector protein downstream of inflammasome (Fig. 1).
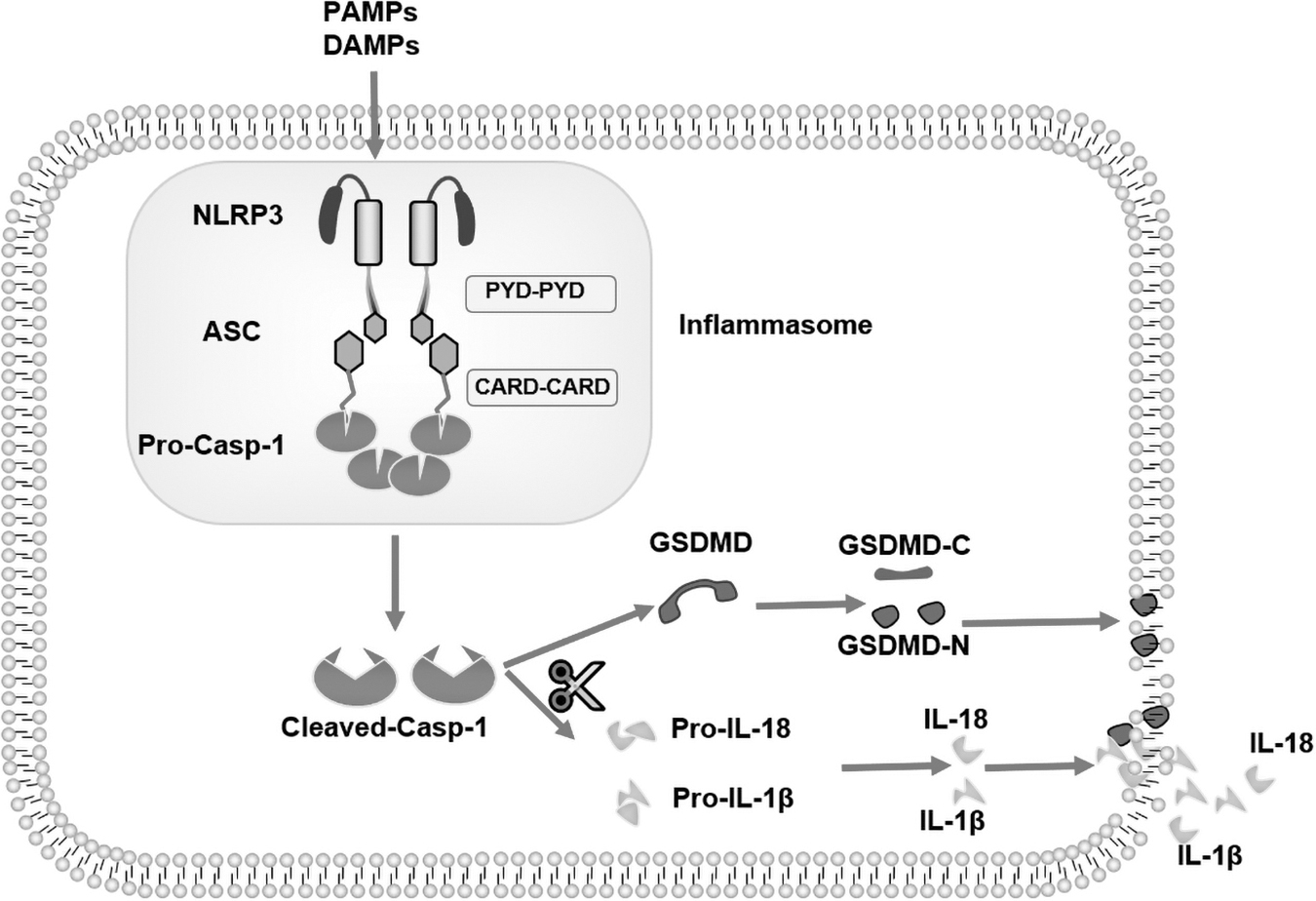
Assembly of NLRP3 inflammasome and its downstream protein molecular pathway. The sensor protein NLRP3 is activated when subjected to stimulatory signals, binds to the adaptor protein ASC and ultimately activates Cleaved Caspase-1. Cleaved Caspase-1 cleaves Pro-IL-1β and Pro-IL-18 to make it mature IL-1β and IL-18, which can induce inflammatory responses. Cleaved Caspase-1 cleaves GSDMD to form the GSDMD-N terminus, which enables the cell membrane to form a honeycomb punch and mediates the effects of pyroptotic death. ASC, apoptosis-associated speck-like protein containing CARD.
More than ten inflammasomes have been identified, including NLRP1 (Martinon et al., 2002), NLRP2, NLRP3 (Agostini et al., 2004), NLRP6, NLRP7, NLRP12 (Tuladhar and Kanneganti, 2020), NLRC4 (Damiano et al., 2001), NLRC5, RIGI, AIM2 (Brunette et al., 2012), IFI16, Pyrin/TRIM20 (Aksentijevich et al., 1997; Yu et al., 2006), and so on. In broad outline, NOD-like receptor (NLR) can be divided into two categories according to the different effects after recognizing PAMP and DAMP. One group is mainly NLRP3, and the other members are NLRP1, NLRP6, NLRC4, and NLRC5.
After activation, they can recruit downstream ASC and pro-Caspase-1 to form classical inflammasome, and promote the maturation and activation of IL-1β and IL-18. IL-1β and IL-18 can localize pathogens and further activate phagocytes. Phagocytes secrete and release more cytokines, phagocytosis invading pathogens. The other group includes NLRP12, NLRX1, and NLRC3, which can inhibit the conduction of the Toll-like receptor (TLR) signal pathway and finally inhibit NF-κB, preventing the transcription and expression of inflammation-related genes, to limit the spread of inflammation (Tuladhar and Kanneganti, 2020). The type of inflammasome is determined by receptor proteins (Wen et al., 2021).
Receptor proteins are divided into two categories: NLR family receptor proteins and non-NLR families. The receptor proteins of the NLR family can be subdivided into receptor proteins containing the PYD domain and receptor proteins containing the CARD domain. The PYD domain on the receptor protein binds to the PYD domain of ASC to form homotypic PYD-PYD interaction. CARd, another domain in ASC, can bind to the CARD domain of effector protein Caspase to form homotypic CARD-CARD interaction. In this way, ASC acts as a connector or bridge to successfully connect receptor proteins and effector proteins (Srinivasula et al., 2002; Jin and Flavell, 2010) (Fig. 1). The NLRP3 receptor protein reviewed in this article is one of the most studied and mature members of 22 human NLR families. The expression of NLRP3 protein is the rate-limiting step of NLRP3 inflammasome activation.
The activation of NLRP3 inflammasome involves two consecutive steps, which are triggered by two signals. The initial step is triggered by the first signal, when cells are stimulated by risk factors, transcription and expression of pro-IL-1β and NLRP3 precursor proteins can be induced by the NF-κB pathway.
The second signal triggers multiple signaling pathways, including reactive oxygen species (ROS) production, lysosomal damage, and the potassium outflow, which induce the assembly of NLRP3 inflammasome (Mangan et al., 2018). When stimulated by agonists, the receptor protein NLRP3 finally activates Caspase-1 through the above interactions, inducing Caspase-1 self-cleavage and activation (Franchi et al., 2009; Tschopp and Schroder, 2010).
As a component of innate immunity, NLRP3 inflammasome plays a critical role in immune response and disease occurrence. A moderate inflammatory response is a defense mechanism induced by dangerous stimulation. However, when the NLRP3 inflammasome is overactivated, it can produce excessive IL-1β and IL-18, resulting in an excessive inflammatory response, which induces cell death and leads to disease (De Nardo and Latz, 2011; Abderrazak et al., 2015; Haneklaus and O'Neill, 2015). Therefore, it is particularly critical to close the innate immune response in time and prevent the overactivation of inflammasome.
Post-translational modification (PTM) of proteins refers to a covalent process that proteins undergo during or after translation, that is, modification groups are added to one or several amino acid residues or groups are cut off by proteolytic hydrolysis to change the properties of proteins. The more common PTMs mainly includes phosphorylation, ubiquitination, acylation (including acetylation), methylation, and glycosylation (Liu et al., 2016). Among them, ubiquitination refers that the Lysine at the C-terminal of ubiquitin molecule is connected with the Lysine on the target protein through isopeptide bond under the action of ubiquitin activator (E1), ubiquitin-binding enzyme (E2), and ubiquitin ligase (E3), causing the structural and functional changes of the target protein (Popovic et al., 2014). Among the above three specific enzymes with different functions, E3 ligase plays a role in identifying specific target proteins, which determines the specificity of modification (Caldeira et al., 2014).
The ubiquitin molecule contains seven internal Lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) and one N-terminal methionine residue (M1). Ubiquitination can be classified into monoubiquitination, multi-monoubiquitination, and polyubiquitination, depending on the number and topology of ubiquitin molecules attached to the protein substrate. Polyubiquitination includes homo-polyubiquitination and hetero-polyubiquitination (mixed ubiquitination). Homo-polyubiquitination can be subdivided into eight subtypes depending on the Lysine residues mentioned above, and the branched ubiquitin chain is a special linkage of the mixed ubiquitination (Hershko and Ciechanover, 1998; Komander and Rape, 2012; Yau and Rape, 2016) (Fig. 2, taking K48- and K63-linked as examples).
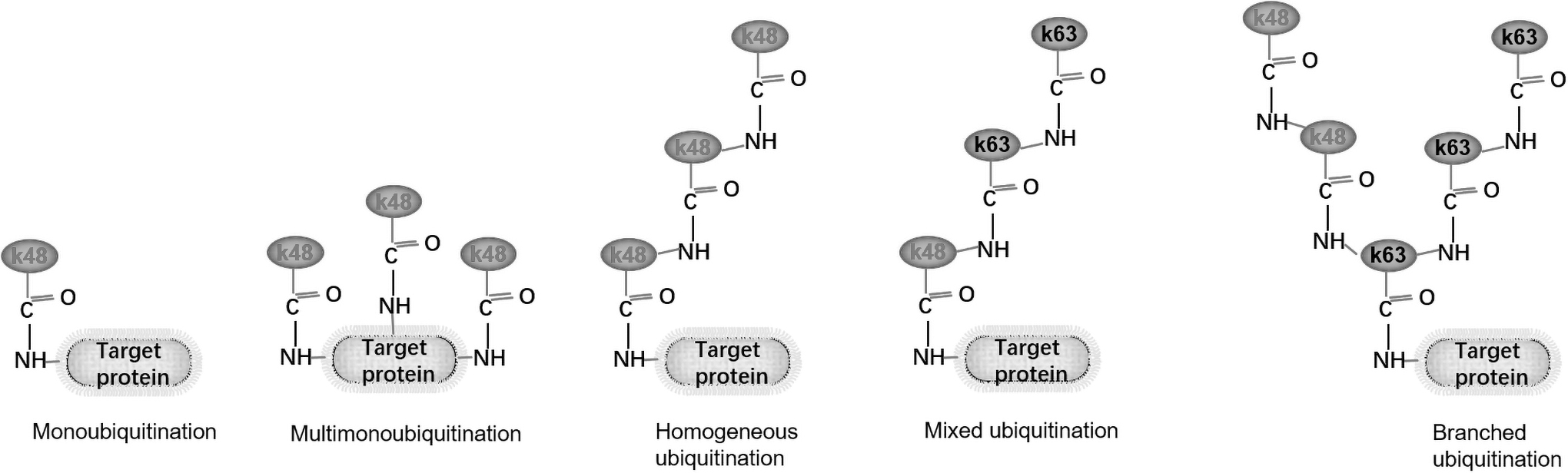
K48- and K63-linked are used as examples for the linkage form between a ubiquitin molecule and a target protein. Depending on the number and topology of ubiquitin molecules linked to the protein substrate, the above follower's monoubiquitination, multi-monoubiquitination, homo-polyubiquitination, mixed ubiquitination. The branched ubiquitination by a special linkage mode of mixed ubiquitination.
At present, the two ubiquitination modification types connected by Lys48 (K48-) and Lys63 (K63-) are the most widely studied. The connection mode of the Lys48 chain can be recognized by the proteasome and used as a degradation signal (Ciechanover and Stanhill, 2014; Stankovic-Valentin and Melchior, 2018). K63 chain or M1 chain is mainly used to establish or dismantle signal complexes, including DNA repair, innate immune response, and NF-κB signaling pathway and other pathways (Yau and Rape, 2016; Rittinger and Ikeda, 2017).
Therefore, ubiquitination plays a critical role in protein localization, metabolism, function, regulation, and degradation, and then participates in the regulation of almost all life activities such as cell cycle, proliferation, apoptosis, differentiation, metastasis, gene expression, transcriptional regulation, signal transmission, damage repair, inflammation, and immunity (Gushchina et al., 2018; Yang et al., 2020; Shen et al., 2021b). Tripartite motif (TRIM) protein is one of the largest subfamilies of E3 ubiquitin ligases. More and more evidence shows that TRIMs play a unique and key role. Their imbalance can lead to the occurrence and development of a variety of diseases, including cancer, immune diseases, and developmental disorders (Hatakeyama, 2017).
Structurally, the conservative TRIMs consist of a RING finger domain (R), one or two B-box domains (B), a coiled-coil domain (CC), and a highly diversified C-terminal domain. The C-terminal includes more than ten different regions, such as COS, PRY, SPRY, acid-rich region (Acid), and so on. Therefore, the main difference between members lies in the number of B-Box and the composition of the C-terminal domain. The RING domain is mainly responsible for transmitting E3 ligase activity to TRIMs, and the complex C-terminal region can mediate the recognition of substrate proteins (Esposito et al., 2017; Hatakeyama, 2017).
There have been excellent reviews on the roles of TRIMs in innate immune responses mediated by TLRs, retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), and cytosolic DNA-sensing receptors, respectively (Yang et al., 2020). In recent years, increasing studies on the regulation of TRIMs against the NLRP3 inflammasome signaling pathway in the innate immune response were reported (Figs. 3 and 4). This article will review the relevant research progress to help us further deepen our understanding of the function of TRIMs and provide some references for guiding the discovery of treatment methods in the future.

Promoting role of TRIMs in NLRP3 and inflammasome pathways. TRIMs are involved in the promotion of the NLRP3 inflammasome signaling pathway by activating the NF-κB/NLRP3 axis, promoting ROS generation, activating members of the inflammasome (NLRP3, ASC, and Pro-Caspase-1), and promoting the formation of GSDMD-N higher-order oligomers, respectively. ROS, reactive oxygen species; TRIM, tripartite motif.

Inhibiting role of TRIMs in NLRP3 and inflammasome pathways. TRIMs upregulate antioxidant protein expression and downregulate ROS generation through the Keap1/Nrf2/ARE pathway, which leads to the inhibition of NLRP3 activation. TRIMs negatively regulate inflammasome activation through the ubiquitination pathway by directly binding to inflammasome members. ARE, antioxidant response element; Keap1, Kelch-like epichlorohydrin-related protein-1; Nrf2, nuclear factor E2-related factor.
Detailed Description About the Role of TRIMs on NLRP3 Inflammasome Signaling Pathway
TRIM proteins are involved in the regulation of NLRP3 inflammasome by ubiquitination of different substrates. Relevant results of previous studies are summarized in Table 1.
Tripartite Motif Proteins Act on Substrates or Signaling Pathways to Regulate NLRP3 Inflammasome
ABHD5, autohydrolase domain containing protein 5; ASC, apoptosis-associated speck-like protein containing CARD; CARD9, Caspase recruitment domain family member 9; Keap1, Kelch-like epichlorohydrin-related protein-1; Nrf2, nuclear factor E2-related factor; ROS, reactive oxygen species; SUMOylation, small ubiquitin-like modifier (SUMO) modification; TRIM, tripartite motif.
TRIM14
Previous studies have reported that TRIM14 can locate to the outer membrane of the mitochondria and interact with mitochondrial antiviral signal protein (MAVs) and plays a role in innate immune regulation and immune anti-infection. After viral infection, K63-linked polyubiquitination occurred at the Lys365 site on TRIM14, and NF-κB essential modulator (NEMO) is recruited into the MAV signal pathway to activate interferon regulatory factor 3 (IRF3) and NF-κB pathway (Zhou et al., 2014). Relevant research results further point out that TRIM14 is NF-κB target gene; TRIM14-NF-κB can form a positive feedback loop to amplify inflammatory signals in activated endothelial cells and promote the progress of human atherosclerosis (Huang et al., 2020).
Studies observed that the level of TRIM14 in the hippocampus of rats with cerebral ischemia/reperfusion (I/R) injury was significantly higher than that in the sham operation group. After injection of TRIM14 siRNA into the left lateral ventricle of rats, the expression level of p-NF-κB, NLRP3, ASC, cleaved Caspase-1, IL-1β, and IL-18 in the hippocampus was detected decreased significantly (Xie et al., 2021), indicating that the potential protective effect of inhibiting TRIM14 under the brain I/R injury in rats is partly through the inhibition of NF-κB/NLRP3 signaling pathway, which plays a role in inhibiting the excessive inflammatory response.
TRIM16
TRIM16 does not have the RING domain that most E3 ligase members contain, but TRIM16 still possesses E3 ligase activity (Bell et al., 2012). Previous findings found that cells cultured in vitro secrete TRIM16 in a Caspase-1-dependent manner; IL-1β also promoted the secretion of TRIM16. IL-1β of secretion similarly increased with TRIM16 overexpression. Further studies showed that TRIM16 interacts with pro-IL-1β, pro-Caspase-1, and NALP1 to promote IL-1β of secretion and involves in the innate immune response (Munding et al., 2006; Rathinam et al., 2012). However, TRIM16 significantly downregulated and IL-1β significantly upregulated in gingival tissue samples from periodontitis patients (Aral et al., 2019).
In further experiments with human gingival fibroblasts (HGFs), Fusobacterium nucleatum (Fn) with or without ATP with or without Porphyromonas gingivalis (Pg) significantly downregulated TRIM16 expression; however, Fn with Pg caused an opposing response (Aral et al., 2020b). Further research is needed to explain why these different results occur. The above phenomenon was not observed in human dental pulp cells (HDPCs), as TRIM16 was not detected to be expressed in HDPCs (Aral et al., 2020a). In periodontal disease, IL-1β plays a crucial role in the host response, and periodontal pathogens may contribute to head and neck squamous cell carcinoma (HNSCC) pathogenesis progression by increasing the IL-1β response due to inflammasome dysregulation (Aral et al., 2020c).
Therefore, strict control of the periodontal pathogens to maintain the dynamic balance between TRIM16 and IL-1β in periodontitis is very important.
Kautilya Kumar Jena and his team found that TRIM16 is essential for the assembly of protein aggregate, while exploring the mechanisms by which cells clear misfolded proteins through the autophagy system. At the same time, TRIM16 is a prosurvival protein that utilizes all its tools (autophagy, nuclear factor E2-related factor [Nrf2]–p62, and ubiquitin system) to keep cells healthy under stress conditions. TRIM16 can release Nrf2 from the inhibitory complex by replacing the position of Kelch like epichlorohydrin-related protein-1 (Keap1) in the Keap1-Nrf2 complex.
At the same time, TRIM16 can increase the ubiquitination of K63-linked Nrf2, promote the activation of Nrf2 (Jena et al., 2018), and enhance the nuclear localization of Nrf2. Nrf2 translocated into the nucleus can upregulate the expression of genes containing antioxidant response elements (ARE), such as heme oxygenase 1 (HMOX1), NADPH:quinone oxidoreductase-1 (NQO1), and autophagy receptor protein p62 (Copple et al., 2010; Jain et al., 2010; Komatsu et al., 2010; Lau et al., 2010), down-regulate the production of ROS, and then inhibit the activation of NLRP3. Therefore, some views believe that TRIM16 is an oxidative stress-inducing gene (Python et al., 2009; Hirose et al., 2011). As a positive feedback mechanism, p62 stabilizes Nrf2, leading to further amplification of the effect of Nrf2 (Jain et al., 2010).
In addition, p62 binds to ubiquitinated misfolded proteins and recruits LC3B (a marker protein for autophagosomes) (Klionsky et al., 2021), leading to autophagy-mediated degradation of ubiquitinated proteins/protein aggregates (Pankiv et al., 2007). It follows that TRIM16 plays a relevant role in protecting cells from oxidative/proteotoxic stress-induced toxicity (Jena et al., 2018). Misfolded proteins are also a feature of neurodegenerative diseases not only because of the neurotoxicity they cause but also because of their effects on overactivated microglia and the NLRP3 inflammasome (Wu et al., 2021a), and microglial NLRP3 inflammasome overactivation has been strongly implicated in the pathology of neurodegenerative diseases (Ising et al., 2019; Tejera et al., 2019).
With these findings above, TRIM16 may serve as a critical therapeutic target protein in inflammatory disease associated with misfolded proteins.
TRIM20
TRIM20 is encoded by the human MEFV (innate immune regulator gene, Pyrin) gene, and MEFV mutations are associated with the autoinflammatory disease familial Mediterranean fever (FMF) (French FMF Consortium et al., 1997). Kimura et al. found that TRIM20 connects with autophagy initiation (ULK1 and Beclin1), elongation (Atg16L1), and execution (Matg8s) factor, to form a platform known as precision autophagy (Deretic et al., 2015), which can directly work on target proteins that need to be degraded with the precision autophagy.
Further investigation revealed that either full-length TRIM20 or a TRIM20 construct containing only the PRY/SPRY domain interacts with NLRP3 and mediates NLRP3 degradation by ULK1 and Beclin1, while in IFN-γ- and LPS-treated cells, downregulation of TRIM20 rescues NLRP3 from degradation (Kimura et al., 2015). In addition to NLRP3, other components, pro-Caspase-1 (Chae et al., 2006; Papin et al., 2007) and NLRP1 (Papin et al., 2007), can interact with the PRY/SPRY domain of TRIM20 and negatively regulate inflammasome activation. When pro-Caspase-1 and NLRP1 are co-expressed with TRIM20, they are protected from degradation by bafilomycin A1, an inhibitor of autophagic degradation (Kimura et al., 2015).
However, TRIM20 can also play an active role in inflammatory pathways and, as an innate immune sensor, the PYD domain of TRIM20 has emerged as a major focus of inflammasome regulation due to its potential to bind the cognate PYD domain of ASC (Schroder and Tschopp, 2010; Vajjhala et al., 2014), which upon release of the PYD domain of TRIM20 triggered by some stimuli, interacts with ASC, can induce ASC oligomerization, instead leads to the activation of inflammasome (Komarow et al., 2003; Yu et al., 2007). It follows that TRIM20 exhibits two sides for the regulation of inflammasome under different stimulation conditions.
TRIM21
During pathogen infection, TRIM21 activates immune signaling through recognition of intracellular antibodies. TRIM21 can catalyze the formation of K63- linked ubiquitin chains and stimulate NF-κB transcription factor pathway activation, leading to the production of proinflammatory cytokines (McEwan et al., 2013). Knockdown of TRIM21 in LPS-treated microglia abolished the high expression levels of NLRP3 protein and Caspase-1 protein induced by LPS, accompanied by the suppressed activity of Caspase-1 and reduced TNF-α, IL-6, and IL-1β secretion, and suppressed the expression of p65 and the binding activity of NF-κB-DNA (Xiao et al., 2021). A recent study showed that TRIM21 is a positive regulator of GSDMD. Knockdown of TRIM21, cell death decreased even the NLRP3, NLRC4 inflammasomes were activated (Gao et al., 2021).
TRIM21 gene excision in mice can reduce LPS-induced inflammation and dextran sodium sulfate-induced colitis. TRIM21 interacts with GSDMD through the PRY/SPRY domain to maintain the stable expression of GSDMD in resting cells. In the cells co-treated with LPS/Nigericin, the size and concentration of GSDMD-N oligomer increased with TRIM21 expression. The results suggest that TRIM21 can promote the formation of GSDMD-N high-order oligomer and induce cell death (Gao et al., 2021). It concludes that TRIM21 plays a critical role in GSDMD-mediated cell death. TRIM21 may become a feasible target for the control and treatment of inflammation-related diseases in the future.
TRIM22
Previous findings have shown that TRIM22 can activate NF-κB (Qiu et al., 2013, 2015), while NF-κB plays a critical role in the NLRP3 inflammasome activation after ischemic stroke (Fann et al., 2018). The expression of TRIM22 significantly increased in cortical tissue subjected to middle cerebral artery I/R ischemia in mice.
Treatment of human neuronal HCN-2 cells with oxygen-glucose deprivation/reoxygenation (OGD/R) found that TRIM22 expression was also significantly upregulated, accompanied by elevated Caspase-3 activity and LDH release. Furthermore, the levels of TNF-α, IL-1β, and other inflammatory factors rise; at the same time, the NLRP3 inflammasome was activated. Knockdown of TRIM22 protects neurons against OGD/R-induced apoptosis and inflammatory responses (Kang et al., 2021). The mechanism of anti-inflammatory effect after knockdown of TRIM22 may be the same as the suppression of TRIM14 expression, resulting from inhibition of the NF-κB/NLRP3-axis.
TRIM24
Endometriosis is an inflammasome-dependent disease. The overactivation of NLRP3 inflammasome is involved in the development and progression of endometriosis (Leavy, 2015). Studies have reported TRIM24 as a negative regulator of inflammation (Josset et al., 2012). The TRIM24 expression decreased in ectopic endometrium of endometriosis compared with that in normal endometrium. Co-immunoprecipitation showed that TRIM24 could interact with NLRP3 in human endometrial stromal cells (hESCs). TRIM24 overexpression promotes the ubiquitination of NLRP3 in hESC, and then NLRP3 is degraded by the proteasome system. Experimental data show that TRIM24 can negatively regulate the cell death and migration of hESCs mediated by the NLRP3 inflammasome signaling pathway (Hang et al., 2021).
However, the specific ubiquitination types and modification sites between TRIM24 and NLRP3 need to be further studied.
TRIM25
The treatment of chronic obstructive pulmonary disease found that epicatechin (EC), a flavonoid, can upregulate the expression of TRIM25 in human bronchial epithelial cells. TRIM25 can promote the ubiquitination and degradation of Keap1 and make it unstable by directly targeting the 615 Lysine site on Keap1 (Liu et al., 2020), which leads to the increase of Nrf2 uncoupled with Keap1 and enhances the nuclear localization of Nrf2. Activated Nrf2 binds to Maf protein to form a heterodimer in the nucleus and then binds to ARE to activate the expression of target genes, regulate the transcriptional activity of antioxidant enzymes, and play the role of antioxidant damage (Michael et al., 2010; de Haan, 2011).
The experimental data showed that EC significantly inhibited the activation of NLRP3 inflammasome and reduced the scorch death of human bronchial epithelial cells induced by tobacco smoke extract (Tian et al., 2021). Thus, we conclude that TRIM25 can reduce the ROS levels and negatively regulate the activation of NLRP3 inflammasome by acting on Keap1/Nrf2/ARE pathway.
TRIM28
Small ubiquitin-like modifier (SUMO) modification (called SUMOylation) is a kind of protein PTM similar to ubiquitination. SUMOylation is mediated by the E1 activating enzyme, single E2 binding enzyme UBC9, and E3 sumo ligase. SUMO proteins include SUMO1, SUMO2, and SUMO3. The three can bind to the target protein in the form of covalent binding as a single molecule (SUMO1) or polymerized SUMO chain (SUMO2 and SUMO3) (Gareau and Lima, 2010). SUMOylation exists in NLRP3 inflammasome activation (Barry et al., 2018; Shao et al., 2020). NLRP3 can interact with the E2-conjugating enzyme UBC9. UBC9 subsequently mediates SUMO1 catalyzed SUMOylation of NLRP3 at Lys204, promoting ASC oligomerization, inflammasome activation, and IL-1β secretion (Shao et al., 2020).
TRIM28 is an E3 SUMOylation ligase (Ivanov et al., 2007). Studies have found that the expression of TRIM28 protein upregulated in macrophages treated with LPS. Further studies found that TRIM28 can promote SUMOylation of NLRP3 catalyzed by SUMO1 and SUMO2/3, thereby weakening or hindering the ubiquitination of NLRP3 in the form of K48- linked, resulting in the enhancement of stability and inflammasome activity of NLRP3. This study revealed the correlation and interaction between SUMOylation and ubiquitination in the process of NLRP3 inflammasome activation (Qin et al., 2021). By the way, not all SUMOylation promotes the activation of NLRP3 inflammasome. Another E3 SUMOylation ligase, MAPL, can inhibit the activation of NLRP3 inflammasome through SUMOylation (Barry et al., 2018).
TRIM30
The dysregulated increase of intracellular ROS is a key stimulating factor leading to the activation of NLRP3 inflammasome. Antioxidant stress treatment can prevent the activation of NLRP3 (Martinon et al., 2009; Kelley et al., 2019). After being stimulated by the NLRP3 agonist, downregulation of TRIM30 in bone marrow-derived macrophages (BMDMs) can increase ROS levels and then enhance the activation of Caspase-1 and increase IL-1β secretion. Overexpression of TRIM30 can reduce the production of ROS and downregulate the activation of NLRP3 inflammasome. Similar phenomena were observed in animal experiments. Monosodium urate (MSU)-induced neutrophil flux and the production of IL-1β were significantly reduced in TRIM30 transgenic mice.
However, the results of co-immunoprecipitation showed that TRIM30 did not interact with NLRP3 and NLRC4, nor with ASC and Caspase-1, regardless of the presence or absence of SPRY (TRIM30) and CARD (ASC and Caspase-1) domains (Hu et al., 2010). Therefore, TRIM30, like TRIM25 summarized above, mainly negatively regulates the activation of NLRP3 inflammasome by downregulating ROS levels. However, the mechanism of TRIM30 regulating the production of ROS is still unclear, which is worthy of further discussion and research.
TRIM31
Treated human periodontal ligament fibroblasts (HPDLFs) with LPS and ATP found that IL-1β increased secretion and upregulated expression of NLRP3, while overexpression of TRIM31 can reverse the above phenomenon (Wu et al., 2020). The study of Chen Jin et al. found that the glucose metabolism of TRIM31−/− mice was impaired with insulin resistance; the expression of p-IRS-1/IRS-1 protein increased and the phosphorylation of Akt (Thr308) decreased. At the same time, it also found that there were significant differences in the composition of intestinal flora, accompanied by an increase in serum TNF-α and IL-1β, and the concentration of TNF-α, IL-1β, Caspase-1, and NLRP3 expression was upregulated in cecum (Cheng et al., 2018).
Song et al. found that when TRIM31 is deficient in vivo; it will enhance the activation of NLRP3 inflammasome and aggravate aluminum-induced peritonitis. Further exploration proved that TRIM31 could directly bind to NLRP3 and promote the polyubiquitination of NLRP3 in a K48- linked, followed by promoting NLRP3 to be degraded by the proteasome (Song et al., 2016). However, since NLRP3 has a protective role in inflammatory bowel disease models (Allen et al., 2010), as opposed to the previous one, deficiency of TRIM31 could, in turn, to some extent attenuate the severity of dextran sodium sulfate-induced colitis.
The results of Zhao et al. showed that under LPS induction, Akt was activated and could promote the phosphorylation of the S5 site of NLRP3 and limit the oligomerization of NLRP3, to prevent excessive activation of inflammasome in the absence of the second signal. And the phosphorylation of the S5 site on NLRP3 inhibits TRIM31-mediated NLRP3 ubiquitination at the k496 site and subsequent degradation (Zhao et al., 2020). When NLRP3 is activated by a second signal, protein phosphatase 2 (PP2A) mediates NLRP3 S5 dephosphorylation, which in turn activates NLRP3 oligomerization and activates NLRP3 inflammasome assembly (Stutz et al., 2017). PP2A-mediated NLRP3 S5 dephosphorylation promotes TRIM31-mediated NLRP3 ubiquitination and degradation.
This mechanism may play a role in preventing excessive inflammatory response after NLRP3 inflammasome activation. Zhao et al. further proposed that Akt, TRIM31, and PP2A can jointly regulate NLRP3 protein level and oligomerization tendency, thus setting a strict regulatory threshold for NLRP3 activation (Zhao et al., 2020). However, another study found that the expression of TRIM31 was significantly downregulated in the ischemic brain.
The results show that the deletion of TRIM31 can reduce the ROS levels, maintain the functional homeostasis of mitochondria, then downregulate the level of neuroinflammation caused by NLRP3 inflammasome, and reduce brain injury caused by middle cerebral artery occlusion. In terms of mechanism, TRIM31 was demonstrated to act on the glycolysis and apoptosis regulator (TIGAR) induced by the p53 tumor suppressor gene. TRIM31 promotes the polyubiquitination of TIGAR and causes it to degrade through the proteasome-dependent pathway, which slows down the clearance process of excess ROS, and then produces a series of oxidative stress reactions and aggravates mitochondrial dysfunction after cerebral ischemia (Zeng et al., 2021).
Therefore, from these studies, we can realize that there are also two sides of TRIM31 in the regulation of NLRP3 under different pathophysiological conditions. Targeted TRIM31 may be another critical therapeutic target for the intervention of NLRP3 inflammasome-related diseases.
TRIM33
TRIM33, also known as transcription intermediary factor 1 γ (TIF1 γ), research on TRIM33 in the last decade has mainly focused on its role in tumorigenesis and progression. In different intracellular contexts, TRIM33 functions both as a tumor suppressor and a promoter (Yu et al., 2019). Related studies have shown that TRIM33 can directly bind to the RNA helicase DHX33 in the cytosol, induce ubiquitination at the Lysine218 on DHX33 through K63- linked, then activate DHX33 (Weng et al., 2014). DHX33 is a cytosolic double-stranded RNA (dsRNA) sensor of the NLRP3 inflammasome that, upon activation, induces Caspase-1-dependent IL-1β and IL-18 production (Mitoma et al., 2013).
Meanwhile, activated DHX33 is essential for the formation of the DHX33-NLRP3 signalosome complex. It was found that downregulation of TRIM33 in THP-1-derived macrophages and human monocyte-derived macrophages was able to suppress dsRNA-induced NLRP3 inflammasome activation (Weng et al., 2014). The potential mechanism may be that after the deletion of TRIM33, DHX33 cannot be ubiquitinated and activated.
TRIM40
TRIM40 is known as the gastrointestinal inflammatory regulator. TRIM40 is highly expressed in the normal gastrointestinal epithelia, but downregulated in gastrointestinal cancers and chronic inflammatory lesions of the gastrointestinal tract. Noguchi et al. (2011) found that TRIM40 could enhance the neddylation of inhibitor of nuclear factor kappaB kinase subunit gamma (IKKγ) and inhibit the activity of NF-κB-mediated transcription, then preventing inflammation-associated carcinogenesis in the gastrointestinal tract.
A recent study on IgA nephropathy found that by treating glomerular mesangial cells (GMCs) with IgA1, NLRP3 inflammasome was detected to be activated. On the other hand, TRIM40 transcript levels, as well as protein expression levels, were downregulated. However, when overexpression of TRIM40, the proliferation of GMCs induced by IgA1 inhibited. The underlying mechanism may be that TRIM40 inhibited the activation of the NLRP3 inflammasome by regulating NLRP3 ubiquitination (Shen et al., 2021a).
Previous studies have also found that TRIM40 promotes proteasome degradation of RIG-I and melanoma differentiation-associated gene 5 (MDA5) through K27- and K48-linked ubiquitination, thereby weakening the innate antiviral immune response. Therefore, controlling the activation of RLRs through TRIM40 and inhibiting the expression of TRIM40 will be a promising intervention for autoimmune and viral diseases (Zhao et al., 2017). Thus, TRIM40 has completely different pathophysiological regulatory roles in regulating inflammation and innate immune response.
TRIM59
Studies have found that TRIM59 is highly expressed in lung cancer cell-derived exosomes, and can be directly transferred from cancer cells to macrophages by exosomes. Affinity capture tandem mass spectrometry analysis indicated that TRIM59 interacts with autohydrolase domain containing protein 5 (ABHD5) and directly induces ubiquitination of ABHD5 (Huttlin et al., 2017), leading to its proteasome-dependent degradation. ABHD5 acts as a liposomal co-activator; deficiency of it can induce metabolic reprogramming of macrophages.
Specifically, it manifested by intracellular fat droplet accumulation, lactate secretion increased, leading to ROS-dependent NLRP3 inflammasome activation and secretion of the proinflammatory cytokine IL-1β increased (Ou et al., 2014), ultimately, convert macrophages into macrophages with Pro tumor functions. That generated a TRIM59/ABHD5/NLRP3/IL-1β-positive feedback loop that promotes cancer cell proliferation and invasion (Liang et al., 2020). The TRIM59/ABHD5/NLRP3 pathway seems to play a role in promoting the proinflammatory cytokine IL-1β generation and mediate an important role in macrophage metabolic reprogramming.
TRIM62
Previous studies have shown that TRIM62 can induce NF-κB activation through the TRIF, a branch of the TLR4 signaling pathway (Uchil et al., 2013). The expression of TRIM62 upregulated in microglial cells treated with oxygen-glucose deprivation (OGD). Meanwhile, it was detected that the TRIM62 expression also elevated in the peri-infarct zone after cerebral ischemia in wild-type mice, while mice with knockout of TRIM62 exhibited neuroinflammation alleviation in ischemic brain tissue, ultimately alleviating stroke complications. Cellular experiments revealed that OGD activated TRIM62 expression by enhancing the interaction of TRIM62 with NLRP3, leading to K63- linked ubiquitination of TRIM62 itself.
Adenovirus-regulated overexpression of TRIM62 promotes NF-κB/NLRP3 signaling pathway, and elevated IL-1β and IL-18 transcriptional activity (Liu et al., 2020). In another study of neuroinflammatory responses triggered by ischemic stroke, the triggering receptor expressed on myeloid cells (TREM)-1 was upregulated in microglia after cerebral ischemic injury. TREM-1, an amplifier of the innate immune response and a regulator of inflammation, can activate the downstream proinflammatory pathway Caspase recruitment domain family member 9 (CARD9)/NF-κB and NLRP3/Caspase-1 by interacting with the spleen tyrosine kinase (Syk) (Xu et al., 2019).
It has been demonstrated in previous studies that TRIM62 can bind to the C-terminal tail of the CARD9 to mediate the ubiquitination of Lys125 on CARD9 in a K27-linked manner, then activating CARD9 (Cao et al., 2015). Activation CARD9 can activate NF-κB inflammatory signaling pathways, in turn, activate downstream signaling molecules, and promote inflammatory factor production (Gross et al., 2006; Drummond et al., 2011). Taken together, TRIM62/CARD9/NF-κB/NLRP3 pathway, which was overexpressed in neuroinflammation, can similarly form a positive feedback loop to promote the development of neuroinflammation.
TRIM65
Our previous study found that in LPS-induced lung inflammation, overexpression of TRIM65 could promote ubiquitination of VCAM-1 in a K48-linked (Li et al., 2020). Subsequently, VCAM-1 was degraded by the proteasome system, thereby inhibiting monocyte adhesion and infiltration into tissues during inflammation and playing a role in protecting endothelial cells from excessive inflammatory stress and alleviating lung injury (Li et al., 2020). Tang et al. found that treatment with the NLRP3 agonist MSU could reduce levels of TRIM65 in a time-dependent manner in PMA-differentiated and LPS-priming THP-1 cells.
Further studies have shown that TRIM65 can bind to NLRP3 and ubiquitinate it. Ubiquitination experiments have detected the simultaneous presence of K48- and K63-linked modifications (Tang et al., 2021). Studies have confirmed that when K48 and K63 are linked to NLRP3 simultaneously, the activity of NLRP3 will be inhibited (Py et al., 2013; Kawashima et al., 2017). Therefore, the author's team believes that upregulation of TRIM65 expression can prevent multiple NLRP3-dependent acute inflammation disease in vivo.
Most studies on TRIM65 have focused on tumors, and studies have found that TRIM65 is highly expressed in colon cancer (Chen et al., 2019), bladder cancer (Wei et al., 2018), liver cancer (Yang et al., 2017), as well as endometriosis (Wu et al., 2021b), and can promote tumor progression. On the other hand, the overactivated NLRP3 inflammasome is tightly associated with tumor development (Karki et al., 2017; Das et al., 2020). Studies have also shown the importance of the NLRP3 inflammasome in the pathogenesis of endometriosis (Leavy, 2015).
Whether there is an interaction between the highly expressed TRIM65 and the overactivated NLRP3 inflammasome in these diseases above deserves further exploration. Whether TRIM65 is a compensatory increase to inhibit the activity of NLRP3 or whether TRIM65 promotes the overactivation of NLRP3 inflammasome remains unresolved.
Discussion and Prospect
Together with the findings above, we understand that the TRIMs play a critical role in regulating the NLRP3 inflammasome signaling pathway. Which function is involved in almost all life activities, such as protein transcription regulation, signal transmission, tissue and cell damage repair related to inflammation and immunity. However, what we know about the function of TRIMs is still limited. Findings demonstrate that E3 ligases can interact with each other to form homotypic or heterotypic E3-RING polymers.
This broad and selective pattern of E3-RING dimerization has the potential to promote the combinatorial complexity of the human ubiquitination cascade (Woodsmith et al., 2012). For example, the dopamine D1 receptor (DRD1) can negatively regulate the NLRP3 inflammasome through cAMP. cAMP promotes E3 ubiquitin ligase MARCH7 to ubiquitinate NLRP3 and degrade NLRP3 (Yan et al., 2015). In the study of Tang et al., instead of degrading NLRP3, TRIM65 can ubiquitinate NLRP3 and inhibit the activation of NLRP3 inflammasome (Tang et al., 2021). Employing a yeast two-hybrid system screen and complimentary co-immunoprecipitation revealed that MARCH7 forms an E3-RING dimer with TRIM65 (Woodsmith et al., 2012). Given this, whether the E3-RING dimer formed between TRIM65 and MARCH7 and participated in subsequent ubiquitin modification results in the study of Yan et al. (2015) and Tang et al. (2021) is worthy of further research.
If so, what is the regulatory mechanism of this dimer on the NLRP3 inflammasome? All these deserve further research. In addition, Tang et al. observed TRIM65 was more effective in mediating Lys63- than Lys48-linked ubiquitination of NLRP3. Combined with previous studies, we know that even a member of the same TRIMs plays different regulatory roles on the NLRP3 inflammatory body signaling pathway under diverse pathophysiological conditions.
When multiple ubiquitin linkage modes are present simultaneously, it will be more dominant as to which one ubiquitin linkage, and thus may be determine the overall ubiquitination outcome. This question deserves further investigation. In conclusion, as one of the members of E3 ligases, the importance of the TRIMs in regulating NLRP3 inflammasome in innate immunity cannot be ignored. It will be interesting and meaningful to take TRIMs as relevant targets for research on treatment of inflammatory diseases.
Footnotes
Authors' Contributions
Z.-S.J. and Z.W. conceptualized and designed the original idea. N.-H.D., Z.-X.Z., and H.-T.L. performed a literature search and analysis and wrote the first draft. Z.T., Z.-F.W., X.-Y.L., and W.-H.X. provided valuable feedback and critically revised the work. N.-H.D. generated the figures and revised the draft. Supervision was provided by Z.W. and Z.-S.J. All authors read and approved the final article.
Authors' Confirmation Statement
All authors have read and approved the content, and agree to submit it for consideration for publication in your journal.
Disclosure Statement
The authors declare no conflict of interests.
Funding Information
This work was supported by National Key Research and Development Program of China (2019YFA0801601 to Z.-S.J.), National Natural Science Foundation of China (81670429, 91839103 to Z.-S.J.), International Joint Laboratory for Arteriosclerotic Disease Research of Hunan Province (2018WK4031), Special Funding for Construction of Innovative Provinces in Hunan Province (2020SK2105 to Z.-S.J.), “Double First-Class” project for innovative Group of Basic Medicine, University of South China (to Z.-S.J.), and Hunan Graduate Scientific Research Innovation Project (CX20210958 to N.-H.D.).