
Abstract
Accumulating evidence has indicated that noncoding RNAs (ncRNAs) are involved in doxorubicin-induced cardiotoxicity (DIC). However, the ncRNA-associated competing endogenous RNA (ceRNA)-mediated regulatory mechanisms in DIC remain unclear. In this study, we aimed to systematically investigate the alterations in expression levels of long noncoding RNA (lncRNA), circular RNA (circRNA), microRNA (miRNA), and mRNA in a DIC mouse model through deep RNA sequencing (RNA-seq). The results showed that 217 lncRNAs, 41 circRNAs, 11 miRNAs and 3633 mRNAs were aberrantly expressed. Moreover, the expression of 12 randomly selected transcripts was determined by real-time quantitative polymerase chain reaction to test the reliability of RNA-seq data. Based on the interaction between miRNAs and mRNAs, as well as lncRNAs/circRNAs and miRNAs, we constructed comprehensive lncRNA or circRNA-associated ceRNA networks in DIC mice. Moreover, we performed Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses for differentially expressed genes. In conclusion, these identified ceRNA interactions provide new insight into the underlying mechanism and may be crucial therapeutic targets of DIC.
Introduction
Doxorubicin (Dox), one of the most effective chemotherapeutic agents, has been used for treatment of a wide variety of solid tumors and hematological malignancies, such as breast cancer and lymphoma (Smith et al., 2010; Cardinale et al., 2015). However, its clinical application has been hampered by its acute and chronic cardiotoxic side effects (Dox-induced cardiotoxicity, DIC). Dox-induced acute cardiotoxicity, also called acute onset cardiotoxicity, usually occurs within 2–3 days after receiving high doses of Dox (Hydock et al., 2009).
On the other hand, chronic DIC is typically positively related to the cumulative dose of Dox (Shabalala et al., 2017). In a retrospective study, Swain et al. (2003) found that patients who received a cumulative Dox dose of 400, 550, and 700 mg/m2 were associated with a 5%, 26%, and 48% increased risk of congestive heart failure, respectively. To date, detailed mechanisms responsible for DIC have been widely discussed.
Among the molecular mechanisms of Dox cardiotoxicity, the formation of reactive oxygen species (ROS) associated with oxidative stress generation is generally accepted (Aziz et al., 2019; Avagimyan et al., 2021). Increased ROS generation induces damage to the heart muscle. Furthermore, DIC also involves mitochondrial dysfunction, perturbation in iron regulatory protein, release of nitric oxide, inflammatory mediators, calcium dysregulation, autophagy, and cell death (Renu et al., 2018; Rawat et al., 2021). However, none of them can fully explain the occurrence of cardiotoxicity. An exploration of both strategies and biomarkers for prevention of this disease is necessary and urgent.
Recently, noncoding RNAs (ncRNAs), including long ncRNAs (lncRNAs; >200 nt long), microRNAs (miRNAs; ∼22 nt long), and circular RNAs (circRNAs), with covalently closed-loop structures have become a research focus (Slack and Chinnaiyan, 2019). Previous studies have found that these ncRNAs play a significant role in multiple human diseases, especially cardiovascular disorders (Esteller, 2011; Poller et al., 2018).
Accumulating evidence has suggested that dysregulation of miRNAs is strongly related to the occurrence and development of DIC (Zhao et al., 2018; Gioffre et al., 2019; Hanouskova et al., 2019). Functional studies also proved that restoration or knockdown of certain miRNAs was able to regulate the progression of DIC (Tony et al., 2015; Wang et al., 2015).
lncRNAs have been demonstrated to play an essential part in DIC through interacting with miRNAs or mRNAs (Li et al., 2018; Xie et al., 2018). Although the functional role of circRNAs in DIC has not been systematically studied, circRNAs are reported to take part in the mechanism of DIC. The series of evidence indicates that ncRNAs may function in progression of DIC.
Since the competing endogenous RNA (ceRNA) hypothesis was proposed in 2011, it has been supported by numerous recent studies (Jeyapalan et al., 2011; Salmena et al., 2011; An et al., 2017; Zhou et al., 2019). The hypothesis describes that ceRNAs, such as lncRNA, circRNA, and mRNA, competitively bind to the common binding sites of target miRNAs, also called miRNA response elements (MREs), thereby regulating the expression of miRNA or mRNA. Both lncRNAs and circRNAs can inhibit the functional miRNAs through the ceRNA network, in which they act as miRNA sponges (Tay et al., 2014).
The ncRNA-associated ceRNA networks may play a key role in inflammation, oxidative stress, apoptosis and autophagy, or other biological activities. For instance, the lncRNA MALAT1 sponges miRNA-92a-3p to inhibit Dox-induced cardiac senescence by targeting ATG4a (Xia et al., 2020). Another study found that circRNA Arhgap12 modulates DIC through targeting the miR-135a-5p-ADCY1 axis (Wang et al., 2021), which provides new evidence that ceRNA may reveal the mechanism of DIC.
The potential connection between DIC and ncRNA-related ceRNA networks provides new ideas for the research of DIC. Although there are several studies on the function of ncRNAs and ceRNA networks in DIC, their potential role in the pathogenesis of DIC has not been fully illustrated. In the present study, we used RNA sequencing (RNA-seq)-based transcriptomic analysis to systematically identify differentially expressed lncRNAs, circRNAs, miRNAs, and mRNAs.
This study established circRNA- or lncRNA-associated ceRNA networks in the DIC mouse model and may provide new insight for diagnosis and therapeutic targets in DIC.
Materials and Methods
Animals and experimental design/protocols
Healthy, female C57BL/6 mice, aged 6–8 weeks and weighing 16–20 g, were purchased from the Laboratory Animal Center, Xiangya School of Medicine, Central South University (Changsha, China). All experimental procedures were performed according to the institutional guidelines and approved by the Medicine Animal Welfare Committee of Xiangya School of Medicine (SYXK-2015/0017). Mice were housed in a temperature- and humidity-controlled room with unrestricted access to water and food. The experiment has been approved by the institutional review board (IRB) of Central South University.
A total of 20 mice were randomly and equally divided into two groups with 10 animals in each group. Mice in the Dox-treated group received a Dox intraperitoneal injection (Zhejiang Hisun Pharmaceutical Co., Ltd, Zhejiang, China) every other day for five times (3 mg/kg, cumulative dosage of 15 mg/kg), and mice assigned to the control group were given a considerable dose of saline.
Seven days after the last Dox injection, all mice were anesthetized with pentobarbital sodium and blood and heart samples were collected immediately. We randomly selected three mice from each group for the following tests.
Biochemical assay and echocardiography
The blood samples collected from anesthetized mice were centrifuged at 3000 rpm at 4°C for 15 min to obtain serum. The serum biochemical parameters, including cardiac troponin T (cTnT), creatine kinase (CK), CK-MB (CK-MB is an isoenzyme of creatine kinase [CK]; CK consists of two subunits, M and B, and has three isoenzymes: MM, MB, and BB), and lactate dehydrogenase (LDH), were analyzed using kits with an automatic biochemical analyzer (Abbott Pharmaceutical Co., Ltd., Lake Bluff, IL). The experiments were performed according to the manufacturers' protocols.
Mice were lightly anesthetized with isoflurane in oxygen after the final injection of Dox or saline. Two-dimensional and M-mode echocardiography was performed using the Vevo 2100 high-resolution in vivo imaging system (VisualSonics, Toronto, ON, Canada) to assess cardiac function. Left ventricular ejection fraction and left ventricular fractional shortening were measured and calculated with the VevoStrain software Work Station.
RNA extraction
Total RNA was extracted from the heart tissues using the TRIzol reagent (Takara Bio, Dalian, China) in accordance with the manufacturer's instructions. The concentration of RNA was determined by the NanoDrop spectrophotometer (Thermo Fisher Scientific). We then used agarose gel electrophoresis to assess RNA integrity.
RNA-seq analyses
RNA-seq libraries for miRNAs were constructed according to the guidelines provided with the TruSeq® miRNA Sample Prep Kit, v2 (Illumina, San Diego, CA). Sequencing was performed on the Illumina HiSeq 2500 platform from Genergy Biotechnology Co., Ltd. (Shanghai, China).
Next, we constructed cDNA libraries of circRNAs, lncRNAs, and mRNAs in accordance with the protocol provided with the TruSeq RNA LT Sample Prep Kit, v2 (Illumina). Then, these libraries were sequenced on the Illumina HiSeq 3000 platform. All subsequent analyses were performed using clean reads after removing the adaptor reads and low-quality tags.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses
In this study, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were applied to investigate the potential functions of differentially expressed ncRNAs and mRNAs. The GO analysis is divided into the following: molecular function, biological process, and cellular component (
KEGG is a database resource applied to explore the significant pathways of differentially expressed genes (DEGs) (
Construction of the ceRNA network
We analyzed the significantly different expression levels of ncRNAs and mRNAs between the control group and the DIC model. miRanda (
We calculated the Pearson correlation coefficient (PPC) and significant p-value between mRNA and circRNA (lncRNA) expression, as well as the number of miRNAs combined with the 3′ untranslated region sequence of mRNA and the circRNA or lncRNA sequence. The ceRNA network was built with RNA pairs with PPC ≥0.5 and p-value <0.05. For each ceRNA pair, a hypergeometric test was applied. Furthermore, shared pairs of miRNA-mRNA and miRNA-lncRNA (circRNA) were used to predict the ceRNA score according to the following formula: ceRNA_score = MRE_for_share_miRNA/MRE_for_lncRNA (circRNA)_miRNA.
Based on the analysis of high-throughput sequencing data, we constructed the circRNA-related ceRNA network and lncRNA-related ceRNA network and visualized these networks using Cytoscape software, v.3.5.0 (San Diego, CA). The specific flow chart of construction and analysis is shown in Figure 1.

Whole experimental protocol of the study and specific flow chart of ceRNA network construction and analysis. ceRNA, competing endogenous RNA.
Real-time quantitative polymerase chain reaction validation
RNA (1 μg) was reverse transcribed into cDNA using a PrimeScript RT reagent kit (Takara Bio) according to the manufacturer's instructions. Real-time polymerase chain reaction (PCR) was performed with a QuantStudio™ 5 Real-Time PCR system (Thermo) using SYBR Green I (ABclonal, Wuhan, China) in accordance with the manufacturer's instructions.
Real-time qPCR analysis was used to validate results derived from RNA-seq. The specific quantitative primers were designed and synthesized by RiboBio (Guangzhou, China) and are listed in detail in Table 1. The primers of ACTB (for lncRNA), GAPDH (for circRNA and mRNA), and U6 (for miRNA) were used as endogenous controls.
Primers Used in Quantitative Real-Time Polymerase Chain Reaction
Statistical analysis
GraphPad Prism 7.00 was used to construct histograms. All data are presented as mean ± standard error of mean and compared using Student's t-test. Sequencing data were processed using DEseq software. The expression level of each circRNA, lncRNA, miRNA, and mRNA is represented as fold change using the 2−△△t method on real-time quantitative PCR (qPCR) analysis.
mRNAs and ncRNAs were defined as differentially expressed when p < 0.05 and |log2 (fold change) | ≥ 1. p-Values <0.05 were considered statistically significant.
Results
Evaluation of the DIC mouse model
Echocardiographic examination showed that Dox treatment displayed changes in left ventricular morphology, characterized by poor contracting ability (Fig. 2A). The ejection fraction and fractional shortening in mice in the Dox group decreased significantly compared with the control group (Fig. 2B). Moreover, histological changes of the myocardium determined by hematoxylin and eosin staining were observed to assess the effect of Dox on cardiac structure. Compared with the control group, the Dox group showed karyolysis, disorganization of muscle fibers, and interstitial edema (Fig. 2C).
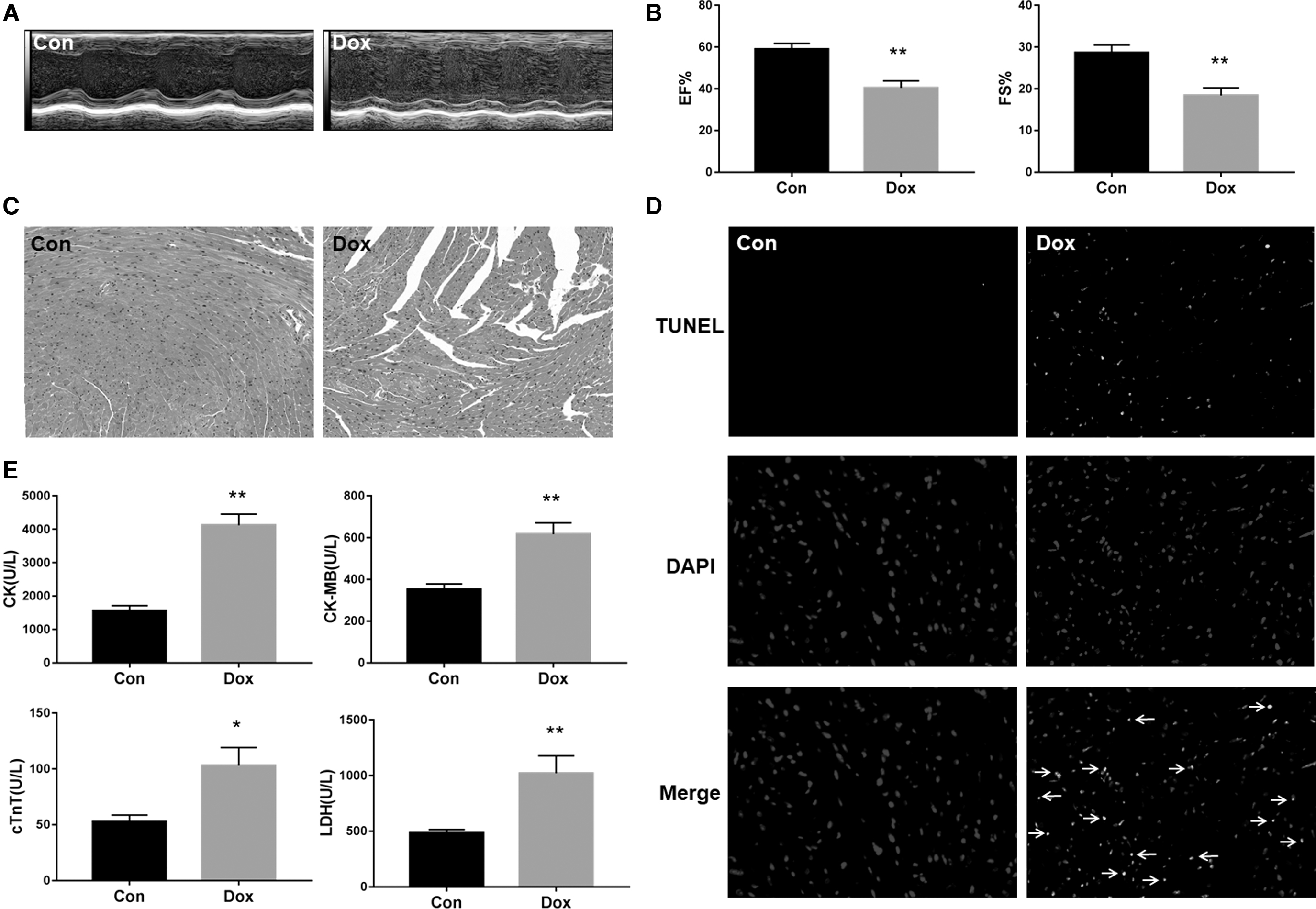
Identification of DIC C57BL/6 mice.
Additionally, an increased percentage of TdT-mediated dUTP Nick-End Labeling (TUNEL) staining-positive cells was observed in Dox-treated mice, indicating the apoptosis of cardiomyocytes (Fig. 2D). Furthermore, the levels of cTnT, CK, CK-MB, and LDH in mouse serum can reflect myocardial injury and strong specificity. We observed that the serum cTnT, CK, CK-MB, and LDH concentrations in Dox-treated mice were significantly higher than those in control group mice (Fig. 2E).
Identification of DEGs
By means of heatmaps with already set screening criteria, differential expression (DE) of ncRNAs and mRNAs in control mice and DIC mice was identified (Fig. 3). Based on transcripts per million, we first identified 41 DEcircRNAs (including 17 upregulated and 24 downregulated DEcircRNAs) (Supplementary Table S1) and 11 DEmiRNAs (including 2 upregulated and 9 downregulated DEmiRNAs) (Supplementary Table S3).
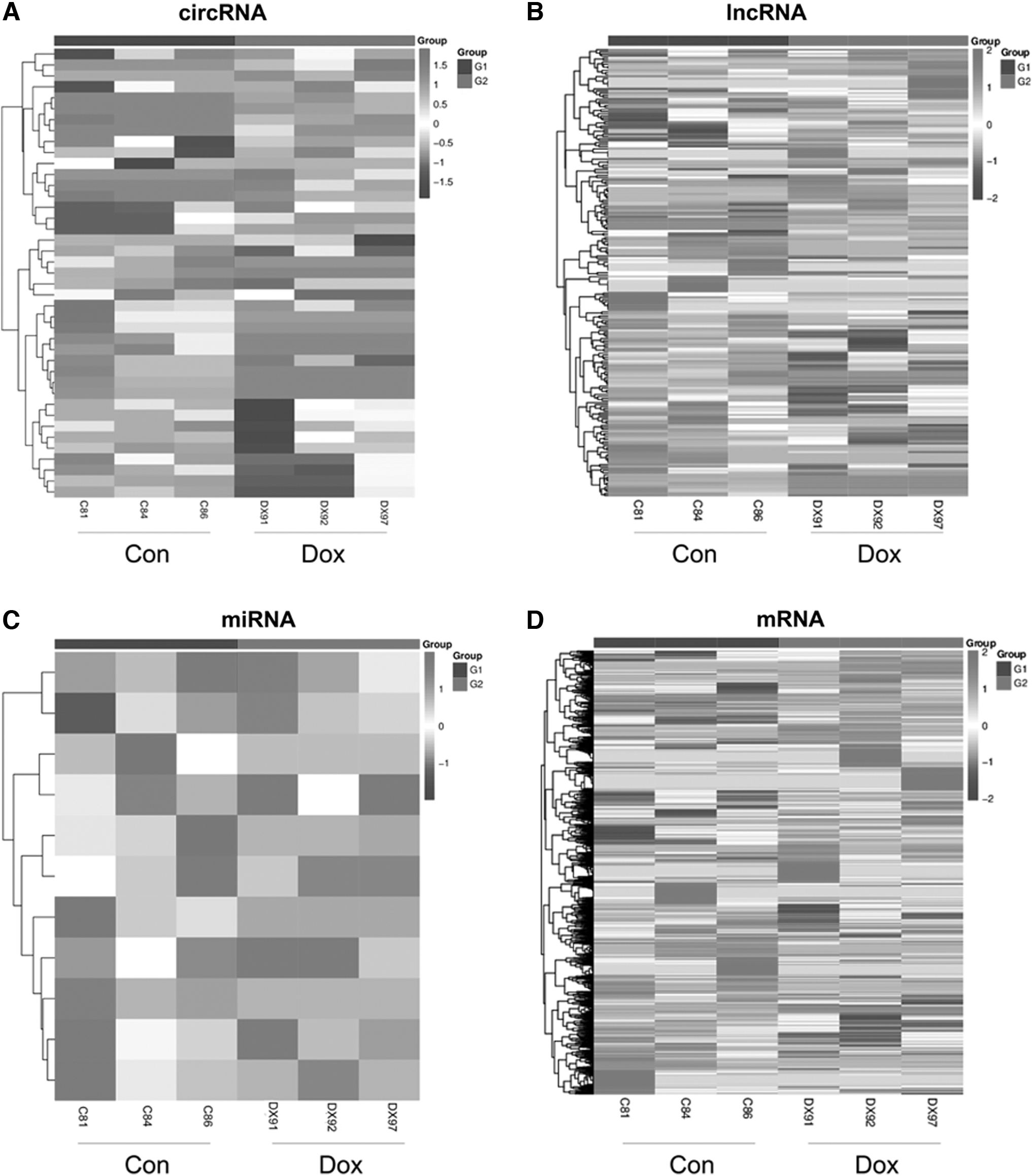
Expression profiles of circRNAs, lncRNAs, miRNAs, and mRNAs. Heatmap of the expression profiles of significantly differentially expressed circRNA
A total of 217 DElncRNAs (including 91 upregulated and 126 downregulated DElncRNAs) (Supplementary Table S2) and 3633 DEmRNAs (including 1904 upregulated and 1729 downregulated DEmRNAs) (Supplementary Table S4) were detected based on fragments per kilobase of exon per million fragments mapped, which is used to estimate the expression levels of lncRNA and mRNA. Among the DEGs, we listed the most upregulated and downregulated one of each mRNA and ncRNA in Table 2.
Statistical Analysis of All Differentially Expressed Noncoding RNAs and mRNAs
circRNA, circular RNA; lncRNA, long noncoding RNA; miRNA, microRNA.
qRT-PCR confirmation
qRT-PCR is usually conducted to confirm the DE obtained from RNA-seq. In the present study, we randomly selected 12 differentially expressed transcripts with three in DElncRNAs, DEcircRNAs, DEmiRNAs, and DEmRNAs, respectively, to test the reliability of RNA-seq data. As shown in Figure 4, the expression levels of selected transcripts determined by qRT-PCR were considerably consistent with the RNA-seq results.

DE levels of lncRNAs, circRNAs, miRNA, and mRNAs were validated by qRT-PCR. The expression levels of lncRNAs (ENSMUST00000228229, Gm16701, and Ino80dos), circRNAs (2:68358814|68410141, 2:76850570|76884197, and 10:116517317|116549175), miRNAs (miR-384-3p, miR-3095-3p, and miR-100-3p), and mRNAs (Ndrg4, G3bp2, and Cyld) detected by RNA-seq were consistent with qRT-PCR results (n = 6–10). DE, differential expression; qRT-PCR, real-time quantitative polymerase chain reaction; RNA-seq, RNA sequencing.
Construction of the ceRNA regulatory network
Next, according to the ceRNA hypothesis, we selected ceRNAs (circRNAs, lncRNAs, and mRNAs) competing for the same MREs of miRNAs and DEmiRNAs to construct an lncRNA- or circRNA-associated ceRNA network (Fig. 5). In the ceRNA network, the DE of mRNAs and circRNAs is generally positively correlated. In total, 33 DEmRNAs, 5 DEcircRNAs, and 7 DElncRNAs were predicted as targets of 7 DEmiRNAs in DIC after being strictly filtered with PPC ≥0.5 and p-value <0.05.
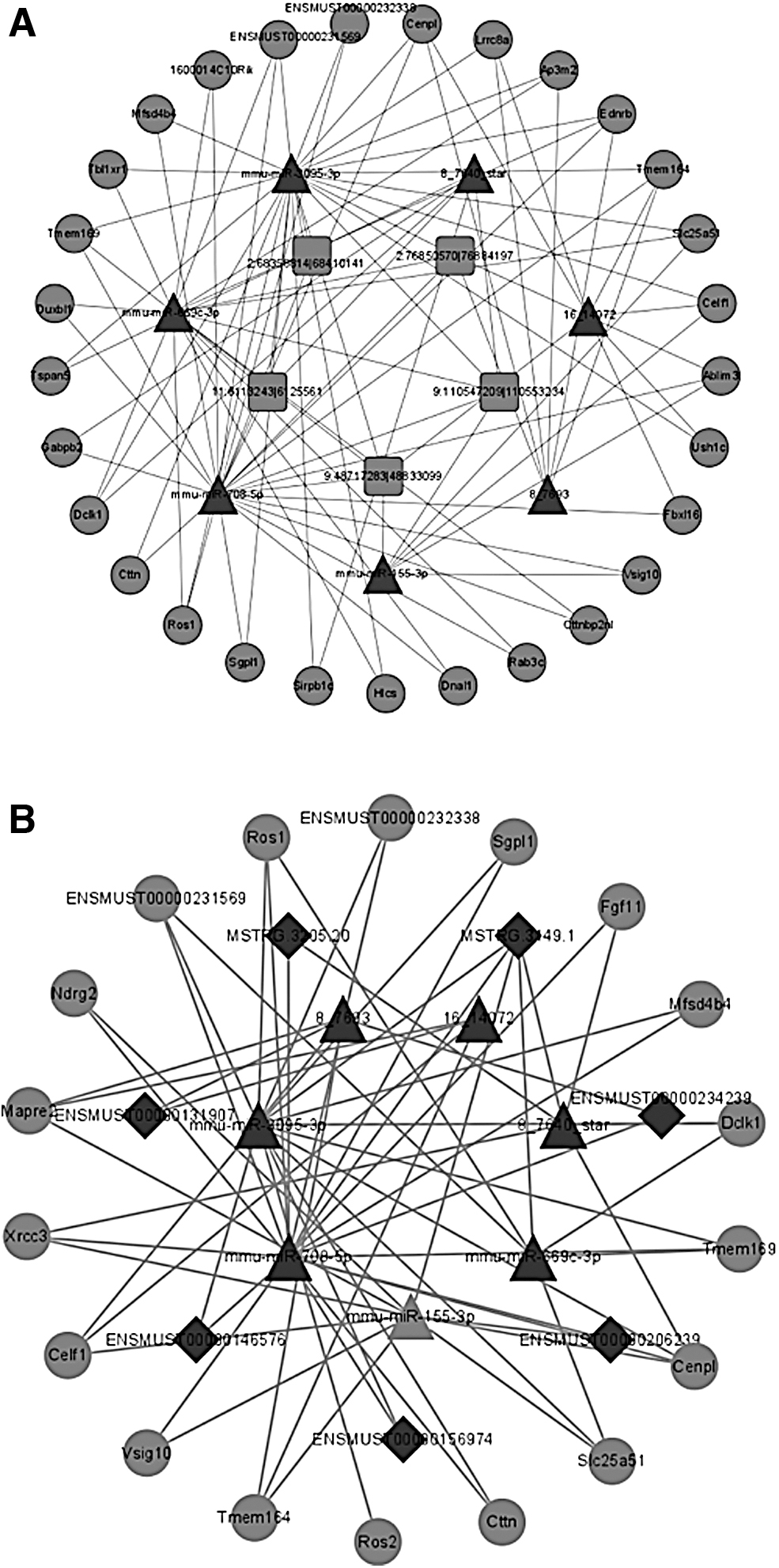
circRNA-associated ceRNA networks in DIC mice and lncRNA-associated ceRNA networks in DIC mice.
Furthermore, we found that there are 29 DEmRNAs, 5 DEcircRNAs, and 7 DEmiRNAs in the circRNA-associated ceRNA network, which contains 73 DEmiRNA-DEmRNA and 21 DEmiRNA-DEcircRNA interactions. The lncRNA-associated ceRNA network contains 17 DEmRNAs, 7 DElncRNAs, and 7 DEmiRNAs, which composes 46 DEmiRNA-DEmRNA and 17 DEmiRNA-DElncRNA interactions. In these two ceRNA networks, miR-708-5p and miR-3095-3p are involved in the most nodes, suggesting that they may act as core regulators.
In addition, miR-155-3p, Celf1, and Ndrg2 have been reported to play an important role in cardiac diseases and Dox treatment. The two ceRNA networks suggest that lncRNAs and circRNAs can absorb miRNAs by sponge action to regulate the mRNA expression. These RNA interactions can be used as a new perspective to explore the potential mechanism of DIC. More details are listed in Tables 3 and 4.
Circular RNA-Associated Competing Endogenous RNA Network in Doxorubicin-Induced Cardiotoxicity
ceRNA, competing endogenous RNA.
Long Noncoding RNA-Associated Competing Endogenous RNA Network in Doxorubicin-Induced Cardiotoxicity
GO and KEGG analyses of DEGs
Based on the mRNAs involved in lncRNA- or circRNA-associated ceRNA networks, we performed, respectively, GO and KEGG analyses. The results showed that DEGs in the circRNA-associated network were significantly enriched in 200 GO terms, of which the top three terms were regulation of multicellular organism growth, outer dynein arm, and biotin-[acetyl-CoA-carboxylase] ligase activity (Fig. 6A). Similarly, 179 GO terms were markedly enriched in the lncRNA-associated network and the top three GO terms were growth, Rad51C-XRCC3 complex, and sphinganine-1-phosphate aldolase activity (Fig. 6C).
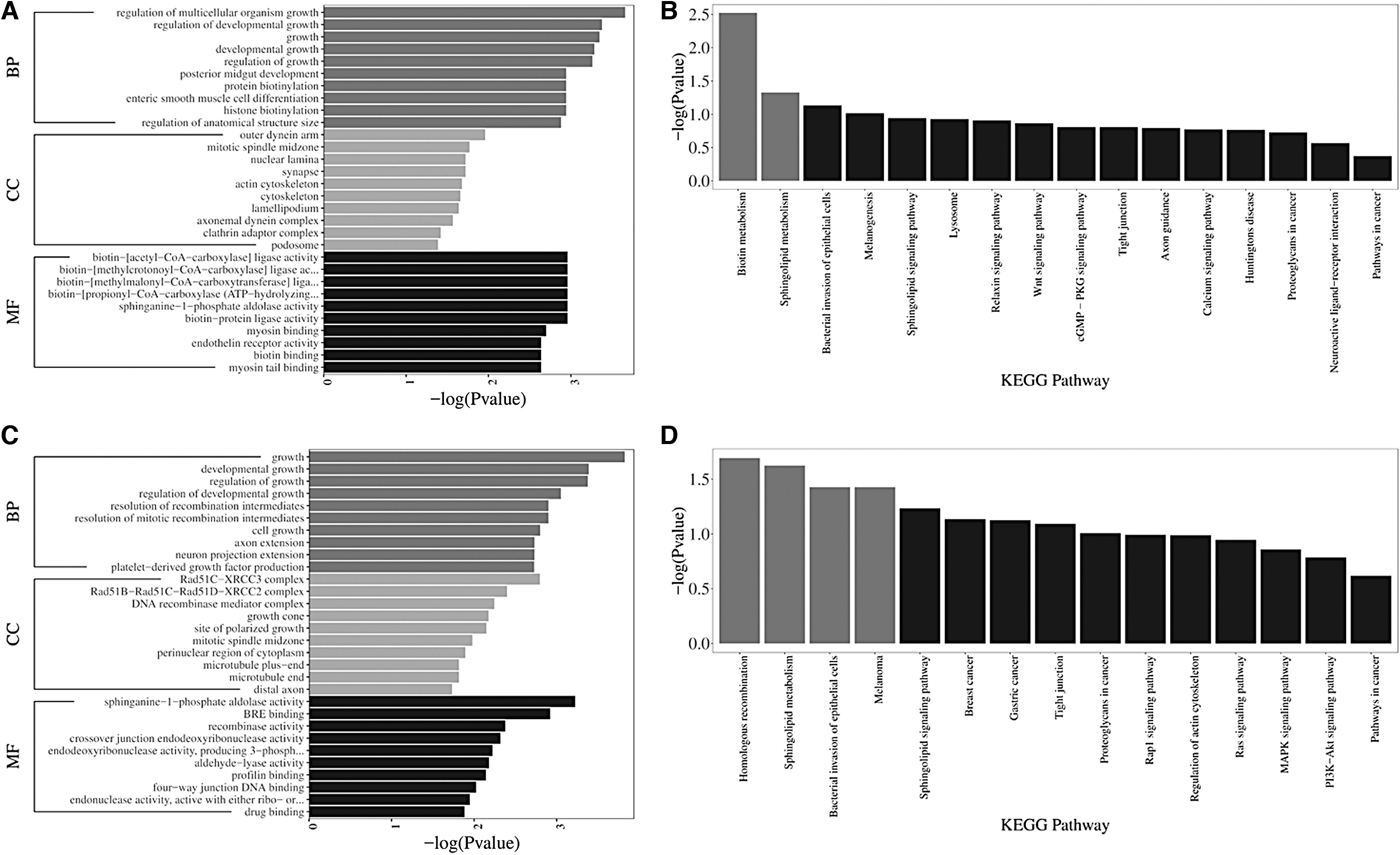
Bioinformatic analysis in the ceRNA network. Data of bioinformatic analysis in the circRNA-associated ceRNA network with GO
KEGG pathway analysis showed that there were two pathways (biotin metabolism and sphingolipid metabolism) and four pathways (bacterial invasion of epithelial cells, homologous recombination, sphingolipid metabolism, and melanoma) enriched in these DEmRNAs involved in the circRNA- and lncRNA-associated ceRNA networks, respectively (Fig. 6B, D).
Discussion
Although Dox is considered as an effective therapeutic option for most cancers, the undesired cardiotoxicity of Dox restricts its optimal application. DIC is usually clinically manifested as cardiac hypertrophy and dilated cardiomyopathy leading to heart failure (Wenningmann et al., 2019). Its cardiotoxic side effects prompted researchers to study detailed mechanisms and develop effective treatments. Nowadays many studies have focused on the emerging role of ncRNA in cardiotoxicity during cancer treatment (Song et al., 2018). However, the role of ncRNA in DIC needs to be further investigated to reveal a new molecular mechanism for DIC.
Disturbance of ncRNA is involved in various complex human diseases by regulating gene expression, especially heart disease (Poller et al., 2018; Cheng et al., 2019; Li et al., 2019; Zhang et al., 2021). Emerging evidence has indicated that ncRNAs participate in cardiomyocyte proliferation, differentiation, and so on (Abbas et al., 2020). Among ncRNAs, miRNAs are the most widely studied in DIC. Studies showed that miR-21, miR-208a, and miR-130a regulate Dox-induced myocyte apoptosis (Tong et al., 2015; Tony et al., 2015; Pakravan et al., 2018).
With the in-depth research on ncRNAs, lncRNA and circRNA are no longer considered as “evolutionary junk” and exert protective or harmful effects during Dox treatment. Inhibition of lncRNA PVT1 and lncRNA CHRF attenuates Dox-induced cardiomyocyte apoptosis (Chen et al., 2018; Zhan et al., 2020). The circRNA, circITCH, as a miRNA sponge, ameliorates DIC through upregulation of SIRT6, Survivin, and SERCA2a (Han et al., 2020).
In this study, we used whole-transcriptome RNA-seq to obtain the differentially expressed RNAs associated with DIC. Our data showed that many dysfunctional ncRNAs, including 41 circRNAs, 217 lncRNAs, 11 miRNAs, and 3633 mRNAs, are involved in DIC. Based on the statistical data of RNA-seq results and ceRNA principle, we established a circRNA-related ceRNA network and an lncRNA-related ceRNA network, containing 5 circRNAs, 7 lncRNAs, 7 miRNAs, and 33 mRNAs, which may be helpful for further analysis of the function of DERNAs in DIC.
Moreover, we used GO and KEGG databases to understand the functions and pathways associated with these screened DERNAs in DIC. GO analysis revealed that these DERNAs were mainly enriched in growth, developmental growth, cytoskeleton, DNA recombinase mediator complex, biotin-[acetyl-CoA-carboxylase] ligase activity, and drug binding, which participated in the pathological process of DIC. Moreover, we found that KEGG pathways such as biotin metabolism and sphingolipid metabolism were closely related to Dox treatment.
It has been reported that biotin conservation in the heart is critical and biotin-related defects are associated with heart disease (Visser et al., 2000). A previous study found that biotin could affect NF-κB-dependent signal transduction (Rodriguez-Melendez and Zempleni, 2003). Modulation of the NF-κB signaling pathway was involved in Dox-induced cardiomyocyte apoptosis (Zhang et al., 2016). Therefore, we speculated that biotin metabolism may participate in DIC. In addition, sphingolipids are a class of bioactive lipids that regulate diverse cell functions. Sphingolipid metabolism is involved in inflammation and oxidative stress.
An emerging study reported that sphingolipid metabolism is of great importance in cardiac tissue in response to ischemic conditions (Hadas et al., 2020). Zhang et al. (2019) reported that astragalus polysaccharides could alleviate Dox-induced cardiomyopathy through sphingolipid and glycerophospholipid metabolism.
In recent years, ceRNA networks have become a popular topic and have been widely and thoroughly studied in many mammalian diseases. In this study, we constructed circRNA- and lncRNA-associated ceRNA networks that are related to the pathogenesis and development of DIC. Among the two ceRNA networks, we found that miR-708-5p, miR-155-3p, Celf1, and Ndrg2 were closely involved in Dox treatment.
miR-708-5p has been demonstrated to exert profound effects in suppressing the oncogenesis of various cancers (Wang et al., 2020; Zhao and Qin, 2020). Interestingly, miR-708-5p is highly expressed in H9C2 cells and neonatal cardiac rodent cells, leading to increased cell proliferation and myocardial regeneration in vivo and protection of myocardial cells from apoptosis by inhibition of MAPK14 expression (Deng et al., 2017). It has been shown that upregulation of miR-708-5p might be involved in the pathogenesis of arrhythmogenic cardiomyopathy (Calore et al., 2019).
In contrast, we found low expression of miR-708-5p during Dox treatment. A previous study demonstrated that miR-155-3p increased the production of proinflammatory mediators, including TNF-α, interleukin (IL)-1, and IL-6, by microglial activation (Zheng et al., 2018). Additionally, miR-155 expression changes with the regulation of cardiac cells. Another study shows that miR-155 can reduce cardiac injury by inhibiting the NF-κB pathway during acute viral myocarditis (Bao and Lin, 2014).
Furthermore, Ling et al. found that miR-155-3p was downregulated during cardiac differentiation of embryonic stem cells (Ling et al., 2017). In addition, miR-155-3p was also downregulated between fetal and adult cardiac remodeling (Yan et al., 2017). The low expression of miR-155-3p is consistent with our study, indicating that it may play an important role in DIC.
Celf1 (CUGBP, Elav-like family member 1) is a key regulator of cardiomyocyte gene expression. Growing evidence shows that increased Celf1 expression is implicated in the cardiac pathogenesis of myotonic dystrophy type 1 and diabetic cardiomyopathy (Wang et al., 2007; Verma et al., 2013). It has been suggested that upregulated Celf1, specifically in cardiomyocytes, plays a role in the transition to decompensation and heart failure (Giudice et al., 2016; Chang et al., 2017).
Inhibition of Celf1 protects cardiomyocytes from acute myocardial infarction-induced injury (Gu et al., 2017). Ndrg2, N-myc downstream-regulated gene 2, is involved in cell apoptosis and survival. Ndrg2, acting as a newly identified mediator of insulin, exerts a cardioprotective effect in myocardial ischemia–reperfusion injury through PI3K/Akt/Ndrg2 signaling (Sun et al., 2013). In the present study, we obtained circRNA-associated ceRNA networks based on strict constraints. Circ 9:48717283|48833099 (up in DIC mice) were ceRNAs of miR-155-3p (down in DIC mice) targeting Celf1 (up in DIC mice).
The circRNA-associated ceRNA network in DIC is highly complex, which needs our ongoing efforts for further exploration.
Conclusions
In summary, we systematically investigated the differentially expressed lncRNAs, circRNAs, miRNAs, and mRNAs in DIC through whole transcriptome sequencing. Moreover, we obtained circRNA- and lncRNA-associated ceRNA networks related to DIC, which provide new insight into the underlying mechanism of DIC.
The role of circ 9:48717283|48833099-miR-155-3p -Celf1 in DIC needs to be further investigated in the future.
Footnotes
Authors' Contributions
B.Z. and W.L. conceived and designed the experiments. Z.X., S.W., and J.H. performed the experiments. Z.X., S.W., and J.L. analyzed the data. Z.X., B.Z., and W.L. wrote the article. All authors contributed to the article and approved the submitted version.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Natural Scientific Foundation of China (nos. 82173911 and 81973406), Fundamental Research Funds for Central Universities of the Central South University (no. 2021zzts1057), Hunan Provincial Natural Scientific Foundation (nos. 2019JJ50849 and 2020JJ4823), Scientific Research Project of Hunan Provincial Health and Family Planning Commission (no. 202113050843), and Bethune Quest-Pharmaceutical Research Capacity Building Project (no. B-19-H-20200622).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.