
Abstract
Abstract
We have recently reported on the impact of CO2 on biomass gasification and its ability to more effectively gasify biomass than steam. Continuing this investigation has led to understanding the impact of heating rates and different reaction environments. This article presents the results from the gasification of various biomass feedstocks. Heating rates were varied from 1 to 100°C min−1 to ballistic rates (∼500°C min−1). Gasification media investigated include H2O/N2, CO2, CO2/N2/H2O, and O2/N2. Global activation energies for pyrolysis were found to be significantly higher than for gasification, whereas those for the grasses were significantly lower than the woods, possibly indicating a catalytic effect during pyrolysis of the high mineral content herbaceous feedstocks. CO2 pyrolysis (110–450°C) activation energy values for lignin, cellulose, and biomass were 22–49, 202–230, and 28–72 kJ mol−1, respectively, and CO2 gasification (500–700°C) values for lignin and biomass were 12–38 and 9–57 kJ mol−1, although cellulose did not exhibit significant mass loss in the gasification interval 500–700°C. Using a least squares fit on the rate of mass loss fraction, the global decomposition reaction during pyrolysis for lignin in either medium was found to be third order, whereas that for cellulose was first order and for the various biomass samples either first or second order. The most significant difference in biomass processing in CO2, when compared with steam gasification, occurred above 750°C where nearly all of the biomass was converted to volatiles with less than 2% ash remaining after CO2 gasification. Only when pure CO2 was used as the gasification medium under a slow heating rate did complete processing of the components to volatile products occur. Gas chromatography analysis has shown the effect of CO2 on product distribution. Data are presented focusing on the relation between gasification medium, feedstock selection, and major chemical species evolution.
Introduction
Development of renewable energy resources such as biomass, as well as technologies such as gasification, which can alleviate global warming issues, offers the opportunity for economic growth in an environmentally responsible manner. They provide the additional benefits of increased economic and political security through local job generation and domestic energy production. Greater control of the volatile evolution products can be achieved by prudent selection of the gasification medium. Steam injection at high gasification temperatures, through the endothermic steam-reforming reaction
has been shown to result in elevated levels of H2 and CO for a variety of carbon-based fuels, including biomass, coal, and municipal solid waste (Minkova et al., 2000; Nehrozoglu, 2004; Senneca, 2007). CO2 injected into the gasification medium can, through the Boudouard reaction,
result in an increase in the CO levels observed. These trends were seen in the gas evolution profiles from all of the hydrocarbon feedstocks studied.
An enhanced microporous network was observed, which resulted from the slow heating of a variety of lignocellulosic feedstocks that were processed under a pure CO2 gasification environment. Compared with the enhanced steam gasification medium, under a CO2 environment a greater processing of the feedstock to volatiles (both permanent gases and condensable species) was found to be possible. Although most of the mass decomposition was observed to occur during pyrolysis, and these low-temperature profiles were found to be nearly independent of %CO2 introduced into the stream, they were highly dependent on the heating rate. The high-temperature profiles for steam vs. CO2 gasification were distinctly different. Only when pure CO2 was used as the reactive gasification medium under a slow (1°C min−1) heating rate, complete processing of the sample to gaseous products was possible. Using CO2 as a valuable thermal treatment medium, while enabling processing at a lower temperature and eliminating the costly expense and energy consumption involved in heating the water used during steam gasification, offers a distinct advantage to CO2 gasification of hydrocarbon feedstocks.
During pyrolysis, the steam introduced for gasification combines with any available CO in the reactor through the WGS reaction, which is thermodynamically favored at lower temperatures,
to produce H2, which can, through direct hydrogenation methanation reactions,
produce a continuous low-level supply of CH4.
At gasification temperatures in the 700–1,200°C range, the dominant reactions that govern biomass steam gasification are the steam-reforming reaction
and the Boudouard reaction
in addition to the cleavage and condensation reactions that break down the biomass lattice structure, the decomposition reactions of the oxygenated minerals releasing O2, and the reaction involving oxidation of the char
Whereas thermal processing in air involves combustion in an oxidizing environment, thermogravimetric analysis (TGA) using N2/H2O or a pure CO2 stream involves a large influent flow that results in gasification under a reducing environment. A wide collection of lignocellulosic decomposition reactions are occurring, and depending on the temperature, O atoms may be leaving the biomass structure through many molecular and radical reactions that include cleavage of the OH, OCH3, C = O, and COOH groups.
TGA studies enable a wide array of potential feedstocks to be studied for comparison regarding their gas evolution characteristics, potential for conversion to volatiles, and ability to differentially process various structural components having a wide range of thermal resilience. They offer the opportunity to derive apparent kinetic parameters that can subsequently aid in process modeling. The thermochemical decomposition behavior of a given feedstock not only depends on its preprocessing but also the nature of the heat and mass transport conditions that it undergoes during thermal treatment. By varying the particle size and total amount of sample undergoing thermal decomposition, information regarding qualitative differences in the nature of surface and volume processing of the feedstock can be ascertained. These studies can provide insight into the means by which the gasification media can react with the char surface to gain access to its inner volume for industrial scale processing.
We have undertaken a detailed study of the gasification products resulting from the thermal processing of grasses, woods, agricultural and forestry residues using steam, air and CO2 as the reactive gasification media. The extent of conversion as well as the H2, CO, CH4, and CO2 evolution from the feedstocks were monitored. The reaction rates were compared as a function of the sample heating rate and we are currently performing a kinetic study comparing the decay rates of cellulose, lignin and various biomass feedstocks as a function of heating rate under both steam/N2 and CO2 gasification media. Although the low-temperature mass decay pyrolysis profiles were nearly independent of percent CO2 introduced into the gasification stream, they were highly dependent upon the heating rate. The high-temperature gasification profiles were distinctly different between CO2 and steam gasification because only when pure CO2 was used as the gasification medium under a slow heating rate, complete processing of the components to volatile products did occur. CO2 offers the opportunity to more completely process the biomass fuel to volatiles. We have shown (Butterman and Castaldi, 2007, 2008, 2009) that CO2 strongly enhances the production of CO at gasification temperatures. This CO can be captured and, through a WGS reaction, used to produce H2 at moderate gasification temperatures.
Analogous to the gasifier CO2 recycle process that we have previously shown (Butterman and Castaldi, 2007) to improve both the carbon conversion and energy efficiency, a similar flue gas looping offers the potential to extract value from what might otherwise be a waste pollutant product. A wood-fired air fed gasification system experiencing a small though significant carbon loss in the ash would typically produce a flue gas rich in CO2 and H2O vapor, with some CO and H2, NO x , and SO2, some CH4 but lower levels of other hydrocarbons, and low levels of soot (unburned C). It is conceivable that once the SO2 and NO x are removed from the flue gas by some sorbent system, recycling of the hot flue gas along with the cool influent for subsequent gasification could aid in the thermal processing of the residual carbon (unburned char) in the ash while offering the opportunity for energy, material, and cost savings.
Experimental Setup
Gasification system
Figure 1 shows a schematic diagram of the gasification test facility in the Combustion Lab at the Columbia University SEAS Henry Krumb School of Mines. It consists of an Instrument Specialists Temperature Programmer Interface/Thermal Analyzer, which can regulate the temperature and heating rate of the quartz furnace in the Dupont 951 Thermogravimetric Analyzer. The carrier flow consists of UHP N2, Bone Dry CO2, and Extra Dry Air whose flow rates are regulated by means of Gilmont GF 1060 rotameters. A kd-Scientific 780-100 syringe pump feeds distilled water into a stainless-steel steam generator that produces slightly superheated steam (∼110–120°C) whose temperature is monitored by an Omega digital E-type thermocouple readout before entering the furnace. The total inlet flow (steam + N2 + CO2) was maintained at 90 mL min−1 except during the 100% CO2 gasification runs when the influent was pure CO2 at a rate of 75 mL min−1. The level of water introduced was maintained at room temperature saturation values while the volumetric % CO2 fed into the line varied from 0% to 60%. Gaseous steam was introduced into the furnace by means of a side arm through which flowed the steam and a portion of the N2 and CO2 remaining after a fraction was diverted for purge flow to prevent deposition onto the TGA electronics. The gasification process was run with excess H2O and CO2 to ensure that the hydrocarbon feedstock was the limiting agent in the steam gasification reactions.
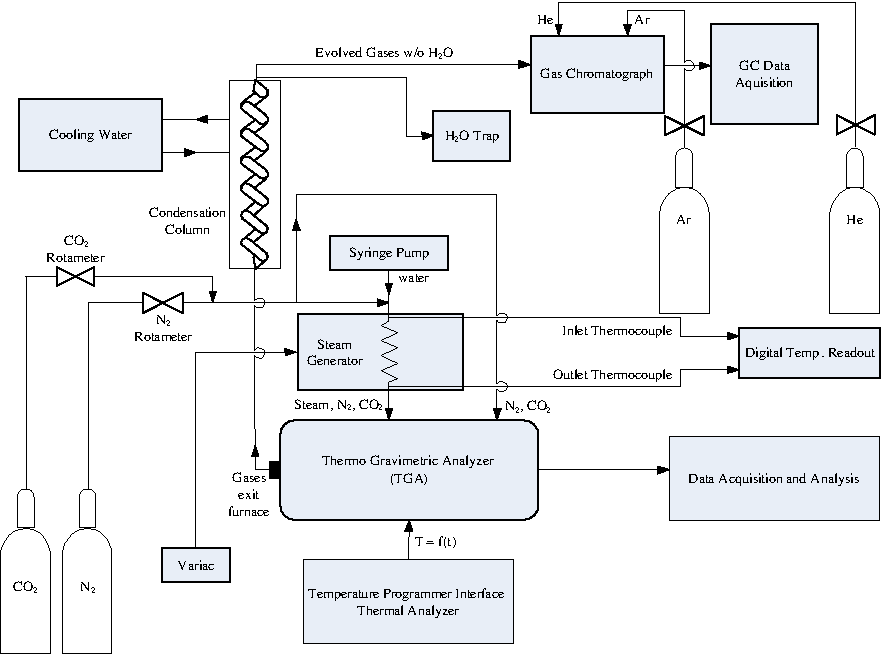
Schematic of experimental apparatus.
The sample is suspended from a quartz rod and sits inside an inert platinum pan. Gas evolution data are recorded as a function of temperature, with the mass decay as a function of temperature displayed graphically in real-time using the programmer interface software. The influent and products of gasification exit the furnace and enter an ice-water condensation column that removes any moisture from the gas evolution products before entering the Agilent 3000A micro-gas chromatograph (Agilent Technologies, Santa Clara, CA). The exit gases leave the furnace and enter the top of the Graham condensation tube in a downflow arrangement while the cooling water enters the bottom in a counterflow pattern permitting condensation at the bottom in the water trap before the gases enter the micro-gas chromatography (GC). During flash pyrolysis, rapid syringe sampling enables gas data acquisition for subsequent GC analysis.
Methodology of gasification testing
Wood samples were prepared by core drilling within planks of untreated wood to produce dry sawdust, whereas the herbaceous feedstocks were air dried, ground by mortar and pestle, and then ball milled. Chemical degradation was minimized by limiting the amount of mechanical processing done on the feedstock. Distinctly different gas evolution profiles exhibited by the green pine needles when compared with the dried pine needles was taken as evidence of this degradation. None of the grasses or needles were water washed, but used as received, to maintain the original mineral content of the sample. Only the beachgrass sample needed careful inspection and pretreatment so that it could be cleaned of all sand grains.
If excessive mechanical working is performed on a feedstock, then the sample becomes significantly hot as to not only lose its moisture but also undergo chemical alteration as evidenced by a color change and a correspondingly different gas evolution profile. The dried pine needles (light brown) were simply air dried and underwent some oxidation in the drying process, whereas the green pine needles were promptly removed from the branches, chopped into small millimeter-sized pieces, and mildly fine ball milled to be processed with minimal heating because of mechanical working and thus minimal chemical degradation as evidenced by the retention of their light green color.
The feedstocks included spruce, white pine, Douglas fir, poplar, sugar maple, red oak, alfalfa, cordgrass, American beachgrass, pine needles, maple bark, blue noble fir needles, wheat straw, Organosolv lignin, powdered crystalline cellulose, and a commercial sample of unprocessed raw mixed wood shavings. The typical size of the samples ranged from 20 to 25 mg. To obtain a representative sample from the feedstock source prepared for TGA, the glass scintillation vials containing ∼5 g of fine wood shavings or a pulverized herbaceous or residue feedstock were well shaken and stirred by spatula before a sample was removed and weighed on the Mettler scale. The sample weights were measured in real-time by the TPI/TA software, with verification by means of a Mettler scale accurate to ± 0.1 mg.
The total influent composed of CO2/N2/H2O introduced into the furnace during gasification was maintained at 90 mL min−1, with the syringe pump regulating the distilled water flow rate, and rotameters regulating the bone dry (99.9% pure) CO2 and UHP (99.999% pure) N2 rates so as to maintain the desired CO2 concentration. Air introduced during flash pyrolysis was varied to see the relative influence of stoichiometry on the gas evolution profiles. The relatively small sample in the TGA pan and the relatively high flow rate of the feed gases result in significant dilution of the gas evolution products detected by the micro-GC.
Results and Discussion
A variety of biomass feedstocks were heated from room temperature to between 840°C and 1,000°C at rates of 1–100°C min−1 under both H2O/N2/CO2 and pure CO2 environments. The mass decay and evolution of H2, CO, CH4, and CO2 during thermal treatment were monitored for each biomass feedstock. GC was used to create the gaseous evolution profiles, TGA to characterize the mass loss during thermal decomposition, calorimetry measurements to calculate the heat content (HHV), and scanning electron microscopy with energy-dispersive X-ray analysis capability (SEM/EDX) to determine the elemental composition of the feedstocks and their gasification residues. As a result, two regimes were identified representing distinct mass decay and gas evolution profiles: low-temperature pyrolysis and high-temperature gasification, whose transition occurs in the vicinity of about 400°C. The introduction of CO2 into the gasification medium produced significantly different gas evolution profiles for H2, CO, and CH4. A distinct CO enhancement and H2 and CH4 depression were observed in both the grasses and woods as the percent CO2 introduced into the influent stream was increased.
We have been able to correlate some of the peaks and troughs in CH4, CO, and H2 evolution with several of the reaction pathways presented in the degradation mechanisms by Jangsawang et al. (2006a, 2006b), Kawamoto et al. (2003), Kawamoto and Saka (2006), Demirbas (2000, 2006), and Banyasz (2001). Cleavage of the lignin structural component results in pyrolysates such as guaiacol and syringol and methyl and methoxy radicals characteristic of the methoxy-phenylpropanoid structure. Cleavage of the cellulose structural component results in pyrolysates such as levoglucosan and other anhydro-glucosaccharides and hydroxyl radicals characteristic of the polysaccharide structure. The distinctly different gas evolution profiles and char structure that are CO2 level dependent indicate both chemical and physical differences in the gasification process when CO2 is introduced. Using CO2 can create a more reactive char and can better access the biomass components by creating a distinctly different char structure. This was clearly visible when the lignin was examined at 860°C after both steam and CO2 gasification. The lignin undergoing steam gasification had a hard, shiny, low porosity surface coating that resulted in less surface area being exposed to the H2O gasification medium. This led to a lower conversion during steam reforming at the elevated temperatures. A very different macropore char structure resulted because of the CO2 gasification of lignin that enabled the CO2 to penetrate the inner volume of the lignin sample with a more complete processing. The macropore structure was not visible following pyrolysis but was observed during gasification at 860°C. By 930°C, though, at this slowest heating rate of 1°C min−1 all of the biomass was processed, leaving no macropore structure in the pan but only a small amount of residue. The influence of CO2 in enabling the more complete processing of a biomass feedstock is visible when comparing the steam gasification of blue fir needles with and without the injection of CO2. Figure 2 shows 0% CO2 injected resulting in a large black char residue after heating the sample from room temperature to 1,000°C at a rate of 10°C min−1. Figure 3 shows the enhanced char burnout using 30% CO2 injected as a co-feed with a resultant small light mineral residue. The most significant reaction responsible for the high-temperature slow heating rate complete processing of the feedstock is the Boudouard reaction between the CO2 and the macropore carbon skeleton occurring at elevated gasification temperatures. Introduction of CO2 enables enhanced thermal processing of carbon-based feedstocks to syngas. Similar to the observations of other investigators, those biomass feedstocks having lower lignin content, such as alfalfa (14%), beachgrass (12%), maple bark (19%), and wheat straw (14%), exhibited characteristically smaller pyrolytic char residues than the high-lignin feedstocks such as the woods and the needles.

Large black char residue from 0% CO2 blue fir needles steam gasification.

Small light mineral residue from 30% CO2 blue fir needles steam gasification.
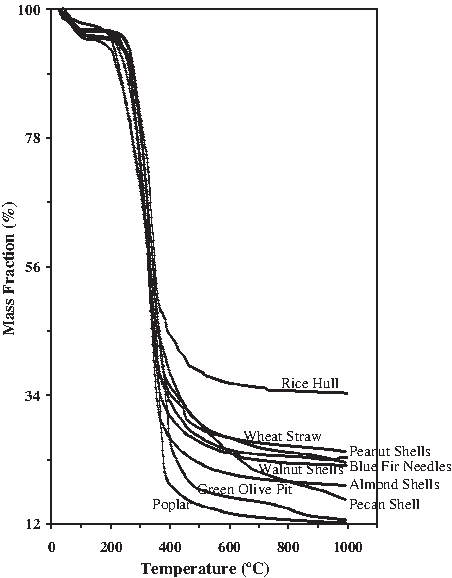
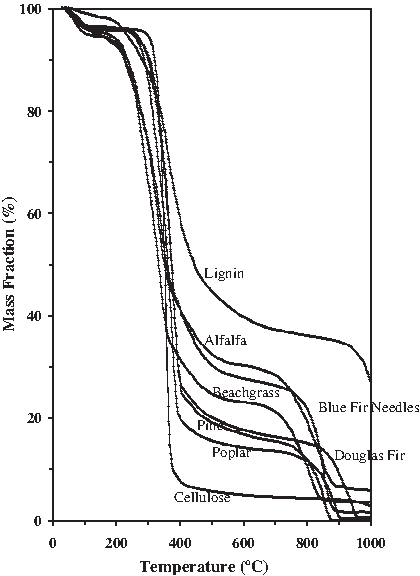
The mass decomposition curve for representative woods, grasses, and agricultural residues undergoing steam gasification is shown in Fig. 4. The largest percent mass loss occurs during pyrolysis between 250°C and 400°C during which time the biomass structural components undergo depolymerization and condensation reactions. As the furnace temperature rises from 400°C to 600°C, during low-temperature gasification, the pyrolytic char undergoes decomposition and decarbonization and the lignocellulosic structure evolves into a carbon lattice skeleton. At high gasification temperatures between 700°C and 850°C, combustion reactions continue the char burnout with the aid of oxygen brought in by the steam and oxygen release from the biomass structure itself. By 1,000°C, all of the biomass samples appear to have completed their mass burnout to residual char and ash. Samples exposed to CO2-enhanced steam gasification had lower residual char fractions. Two distinct mass decay regimes that correlate with the two distinctly different gas evolution regimes can be identified in the decomposition curve where the transition temperature appears to be in the vicinity of 400°C. The mass decay curves are for a slow gasification rate of 10°C min−1 and would be significantly different as discussed by Brunner and Roberts (1980) at higher heating rates. The thermal decomposition of the phenolic and highly crosslinked lignin structure follows a very different set of reaction pathways than that of the polysaccharide cellulose and hemicellulose structures. Lignin decomposition begins much earlier at about 225°C and takes much longer until about 625°C to complete, whereas cellulose decomposition begins later at about 325°C and occurs rapidly so that by 425°C decomposition is nearly complete. The behavior of the structural components is manifested in the resultant decomposition behavior of the biomass feedstock. The various feedstocks showed decomposition rates that were intermediate between that of lignin and cellulose though we can expect that residues or grasses high in alkaline content may also exhibit a catalytic effect not present in the decomposition of the pure structural components. This can be seen in Fig. 5 where the mass fraction percent is bounded below by the cellulose curve and above by the lignin curve, with feedstocks high in mineral composition (beachgrass, alfalfa, blue fir needles) exhibiting the earliest onset of thermal decomposition. In Fig. 4 the feedstocks high in mineral content (blue fir needles, green olive pit) also exhibited an earlier onset of pyrolytic decay. At a heating rate of 10°C min−1 from ambient temperature to 1,000°C, the mass fraction dropped to <1% in CO2 but remained >12% in steam. Processing of poplar and blue fir needles in steam resulted in 12.3% and 22.5% residues, respectively, whereas processing in CO2 resulted in only 0.2% and 1.6%, respectively. CO2 offered significantly improved conversion to volatiles at high gasification temperatures. Pyrolytic decomposition begins earlier for the biomass samples when compared with the “pure” structural surrogates, with complete processing occurring for the actual biomass feedstocks between 850°C and 1,000°C.
Mass decomposition curve for steam gasification (10°C min−1, 0% CO2).
Mass decomposition curve for CO2 gasification (10°C min−1, 100% CO2).
To better understand the nature of the solid combustion products, in addition to the volatile species and solid ash residue gasification products, several biomass feedstocks were combusted in a furnace in air at atmospheric pressure for 30 min at 900°C. The two grasses that are particularly high in alkaline mineral content, alfalfa and cordgrass, were observed to yield the highest mass percent combustion residues and to dissolve through the ceramic glaze of the furnace crucibles. A summary of the classification and structural composition of the biomass feedstocks as well as the results of the furnace and calorimetry measurements are given in Table 1. The feedstocks are not pretreated to remove either moisture or mineral impurities. Calorimetry measurements involve complete combustion of the raw feedstock in a high-pressure pure O2 environment with condensation of H2O as the combustion chamber cools in the water jacket. For those feedstocks whose higher heating value (HHV) was not calculated using the calorimeter, database values are presented from either the Phyllis biomass database of the Energy Research Center of the Netherlands (ECN; 2009), Biomass Energy Foundation (BEF; 2009), Southwest Consortium for Environmental Research and Policy (SCERP; Stewart and Silcox, 1999), or India Institute of Technology (IIT; Safi et al., 2004). Calorimetry measurements agree with these and the Oak Ridge National Laboratory (ORNL; 2009) and European Agriculture and Forestry Biomass Network (EAFBN; 2009) database values. The lignocellulosic structural composition was taken from the various databases including the U.S. Department of Energy Biomass Program (2009).
HHV, higher, heating value; ECN, Energy Research Center of the Netherlands; BEF, Biomass Energy Foundation; IIT, India Institute of Technology; SCERP, Southwest Consortium for Environmental Research and Policy.
The distinctly different gas evolution profiles and char structure that are CO2 level dependent indicate both chemical and physical differences in the gasification process when CO2 is introduced. Using CO2 can create a more reactive char and can better access the biomass components by creating a distinctly different char structure. This was clearly visible when the lignin was examined at 860°C after both steam and CO2 gasification. The lignin that underwent steam gasification (Fig. 6) had a hard, shiny, low-porosity surface coating that resulted in less surface area being exposed to the H2O gasification medium. This led to a lower conversion during steam reforming at the elevated temperatures. The distinctly different physical mechanism involved in CO2 gasification, aside from the chemical differences observed in the contrasting gas evolution profiles, can be observed in Fig. 7. The macroporous structure that was observed was unique to CO2 gasification. It can help to explain why CO2 is so effective in processing the biomass feedstock during high-temperature thermal processing. Messenbock et al. (1999) examined the enhanced char reactivity of coal char subjected to gasification temperatures using CO2 to facilitate the burnout of the thermally resistant unreactive surface layer. Their scanning electron micrographs demonstrated a distinctly different pore structure with the development of an extensive microporous network when they used CO2 that was not observed when using H2O instead. Figure 7 shows the lignin processing in CO2, at 1°C min−1 from ambient to 860°C, resulting in a microporous char structure offering greater surface area and access through the intricate network of channels. By 930°C this char was completely converted to volatiles with only the mineral ash residue from the Organosolv lignin remaining in the pan appearing as a thin film in Fig. 8. A very different macropore char structure resulted because of CO2 gasification of lignin that enabled the CO2 to penetrate the inner volume of the lignin sample with a more complete processing. The macropore structure, shown in Fig. 7, was not visible following pyrolysis but was observed during gasification at 860°C. By 930°C, though, at this slowest heating rate of 1°C min−1 all of the biomass was processed, leaving no macropore structure in the pan but only a small amount of residue as seen in Fig. 8. Heat and mass transport limitations can significantly alter both the chemical and physical processing of a carbon-based feedstock. Sufficient time and thermal energy is necessary for the CO2 to access the inner volume of the feedstock. At rapid heating rates (∼100°C min−1), CO2 can undergo surface reactions but is incapable of accessing the inner volume to create a highly porous structure, volatiles are unable to escape, and a balloon-like structure develops with a hard impervious outer shell as seen in Fig. 9.

Lignin, H2O/N2, 1°C min−1, 22–860°C.

Lignin, CO2, 1°C min−1, 22–860°C.

Lignin, CO2, 1°C min−1, 22–930°C.

Lignin, CO2, 100°C min−1, 22–1,000°C.
When compared with steam gasification, a strikingly different physical mechanism is involved in CO2 gasification macroscopically visible in Fig. 7. Messenbock et al. (1999) also observed this extensive microporous network to occur when they used CO2 but not when using H2O instead. The macroporous structure can help to explain why CO2 is so effective in accessing the char, and so processing the carbon feedstock, during high-temperature thermal treatment. Although the reactive medium plays a significant role in the high-temperature processing, the heating rate is most significant at the lower pyrolysis temperatures. Brunner and Roberts (1980) studied the char properties resulting from cellulose pyrolysis. They found the yield in micropore volume and surface area of slowly heated cellulose to be several times as large as that resulting from a high heating rate. For the processing of lignin in CO2 at a slow heating rate of 1°C min−1 from ambient to 860°C, a macroporous char structure resulted, offering greater surface area and access through the intricate network of channels. Through subsequent heating to 930°C, this char was completely converted to volatiles with only the mineral ash residue from the Organosolv lignin remaining in the pan, seen as a thin film in Fig. 8.
Distinction between surface and volume reactions during high-temperature processing of lignocellulosic feedstocks is more clearly seen when examining the processing of the thermally resistant structural component lignin. Even at very slow heating rates with long residence times, tar forming gasification reactions tend to create a viscous, glassy coating with trapped gases that can undergo secondary char-forming reactions during processing in H2O/N2, as seen in Fig. 6. In contrast, at very slow heating rates in CO2 the pyrolytic char develops a successively more porous structure as the CO2 gains access to the inner volume channels, as seen in Fig. 7. Although initially the surface area increases, once the lignin has become a highly porous sponge-like structure, the surface area decreases while the inner volume increases. CO2 continues to react with the char for complete conversion to volatiles above 900°C, as seen in Fig. 8.
Through the use of the scanning electron microscope with energy-dispersive X-ray spectroscopy capability of the Materials Research Science and Engineering Center at Columbia, the chemical composition of the carbon-based feedstocks and their gasification residues were studied. Table 2 presents the weight percent results appearing in the Princeton Gamma Tech software report. The presence of K and Cl can reduce the ash melting temperature and can account for the corrosion and slagging observed during gasification of herbaceous feedstocks. The carbon content of raw woods such as pulverized poplar (91.91% C) was found to be higher than raw grasses such as alfalfa leaf (79.13% C). The steam gasification of beachgrass resulted in a char in which the C and O levels decreased and the mineral levels in the char significantly increased above those of the raw pulverized beachgrass sample. K rose from 4.99% to 27.23%, Ca from 2.45% to 14.25%, P from 1.31% to 8.69%, Fe from 6.23% to 15.35%, and Si from 0.61% to 8.79%. A visible embrittlement of the quartz rod and furnace, attributable to the strong alkaline flux K2O, was reported as evidence of the devitrification by Butterman and Castaldi (2009). These changes are similar to the crystalline phase transformations that occur during the sintering of silica pottery glazes. MgO and CaO are highly efficient high-temperature fluxes that are also responsible for the highly reactive and embrittling character of herbaceous and residue feedstocks. CO2-enhanced steam gasification of biomass results in more highly reactive chars not only due to the gasification medium but also due to catalytic effects resulting from the mineral composition. The CO2 gasification of beachgrass resulted in only a mineral residue, whereas the steam gasification (0% CO2) of beachgrass exhibited a characteristically large carbon char that was coated with a white mineral residue determined by EDX to be mainly comprised of K, Ca, and Fe. The cellulose pyrolysis mechanism developed by Kawamoto et al. (2003) involves a major pathway in which levoglucosan feeds the volatile pyrolysis products into the gasification reactions. This can help explain the thick nonreactive carbon char or mineral ash residue, carbonization or mineralization, observed for varying CO2 input rates. In the Kawamoto mechanism, levoglucosan serves as the intermediate that can either be broken down to low-molecular-weight products with more complete processing and greater gas evolution concentrations in the presence of sufficient oxygen atoms (such as that supplied by the CO2 input), or undergo polymerization reactions to form polysaccharides with high levels of surface carbonization and large quantities of char in the absence of sufficient oxygen from O or CO. Both of the surrogate lignocellulosic structural models were analyzed. A significant level of oxygen, 11.46%, is contained in the cellulose powder, whereas the carbon content of the Organosolv lignin, 90.35%, is comparable to that of the woods.
SEM/EDX is a useful analytical tool for elemental characterization, though it is not as accurate a quantitative measure as is a chemical assay. Although the H peak is too low to be detected in the EDX spectrum and other elements and/or compounds may be volatized by the electron beam (carried away by the vacuum to go undetected), an SEM/EDX analysis enables initial qualitative identification of many mineral impurities and surface features for comparison of biomass samples.
While steam was observed to significantly improve the H2 production at elevated temperatures, CO2 was observed to enable a more complete processing of the lignin component at higher temperatures. Once low-temperature pyrolysis is complete at about 375°C, the cellulose component has undergone nearly all of its conversion to volatiles, whereas a significant portion (∼70%) of the lignin remains. Further thermal and chemical catalytic treatment of the lignin component, the pyrolytic biomass char, can result in the production of aromatics, alkanes (precursors to diesel fuel), benzene derivatives, alcohols, and oxygenated compounds (some of which have been found to be potential enhanced combustion fuel additives) (Wooley and Abatzoglou, 2000; Cortright, 2007; Myers, 2007; Regalbuto, 2007).
By varying the heating rate of the TGA furnace as well as the percent CO2 introduced into the gasification medium, we attempted to find conditions under which we could separate the greatest portion of lignin from the cellulosic biomass components. The intent was to thermally process the cellulosic component at low temperature and then seek to find ways of thermally and possibly chemically treating the residual lignin component that was more resistant to thermal degradation. While polymerization, ring opening, and condensation reactions characterize thermal hydrocracking, more selective cleavage of the characteristic lignitic ether bonds rather than the aromatic rings can be achieved with catalytic hydrocracking. A combination of both platforms can process the polysaccharide to volatiles at low temperature while optimizing the percent of lignin in the pyrolytic char that remains available for subsequent conversion.
The influence of CO2 on the gas evolution products during steam gasification of biomass feedstocks is shown in Figs. 10–12. A constant 90 mL min−1 of influent composed of N2 and a saturation level of H2O introduced into the TGA furnace reactor as steam, along with varying percents of CO2, were used to gasify the beachgrass and poplar. The heating rate was 10°C min−1 and the temperature ranged from ambient to 1,000°C. As reported by Butterman and Castaldi (2007, 2008, 2009), CO2 has been shown to increase CO production and this enhancement, seen in Fig. 10, becomes significant above 600°C when the Boudouard, steam gasification, reverse WGS (RWGS), and char combustion reactions all favor the increase in CO concentration with temperature. CO2 has been shown to decrease H2 production, as seen in Fig. 11, with the depression becoming significant at the higher gasification temperatures above 600°C. This H2 depression may be due to the high concentrations of CO resulting from the Boudouard and char burnout reactions causing a reverse shift in the steam-reforming reaction. Similarly, CO2 results in CH4 evolution depression, as seen in Fig. 12, due in part to the decreased availability of H2 during gasification, which could enter into the methanation reactions. Increased availability of CO2 would favor the RWGS reaction even at low temperatures resulting in decreased H2 production as well as favor a reverse shift in the highly exothermic methanation reaction (4) resulting in decreased CH4 production due to CO2 injection. During cellulose pyrolysis, hydroxyl radicals are cleaved off of the structure and readily react with CO, CH4, and other hydrocarbon and radical species available in the gasification medium. The OH radical can react with available CO to produce CO2 and a H atom. These H atoms can readily enter into reactions with the CH3 radicals cleaved off of the lignin structure. Injection of CO2 would tend to favor OH reactions with CH4 rather than CO during pyrolysis, which would help to explain the CH4 depression that begins at about 300°C at the onset of pyrolysis.
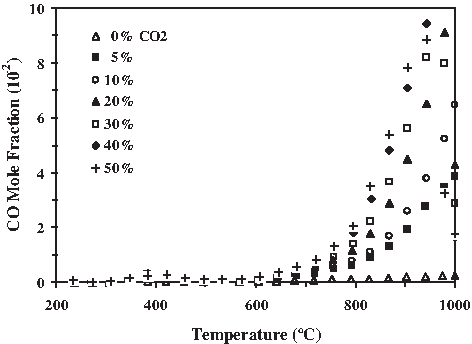
CO evolution enhancement from beachgrass using CO2 (steam-to-carbon, S/C = 18) for T > 750°C.
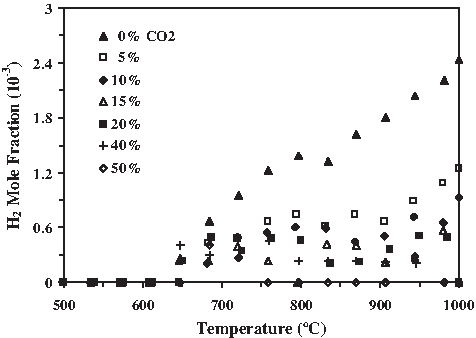
H2 evolution depression from poplar using CO2 (S/C = 21) for T > 700°C.

CH4 evolution depression from beachgrass using CO2 (S/C = 5.5) for T > 300°C.
Several biomass feedstocks were examined and it showed similar gas evolution behavior during high-temperature CO2-enhanced gasification. The four gases that were monitored included CO, H2, CH4, and CO2. Gas evolution profiles for a variety of lignocellulosic fuels that included woods, grasses, and agricultural and forestry residues showed gas concentrations that were highly dependent on the level of CO2 in the influent. All of the gas evolution profiles could be partitioned into a low-temperature and high-temperature regime whose transition occurred at about 400°C, analogous to the behavior demonstrated by the mass decomposition profiles. Although the lignocellulosic structure of biomass is significantly different than that of the carbon lattice of coal or the polymer matrix component of municipal solid waste (MSW), at high temperatures the reduced carbon skeleton behaves similarly for all of these hydrocarbon feedstocks. Though CO2 and CH4 production is favored at lower pyrolysis temperatures, the thermodynamically favored species at high gasification temperatures are CO and H2. When radical reactions become significant, above 250°C, H and CH3 radicals formed from H abstraction and β-carbon scission reactions can begin to form CH4. A lignin β-aryl phenolic elimination mechanism proposed by Kawamoto and Saka (2006), occurring at pyrolysis temperatures from about 250 to 400°C, results in the release of OH radicals. In this temperature interval, cellulose also cleaves OH radicals that branch off of the linear chain. These OH radicals combine with available CO to produce CO2 and generate new H radicals. This accounts for the low levels of CH4 preceding transition to the gasification regime at about 400°C. At about 400°C, alternative lignin degradation pathways proposed by Kawamoto and Saka (2006) become available. These involve β-aryl and β-ether nonphenolic cleavage in which OH radicals are not produced, which would otherwise have consumed the CO needed in the methanation reactions. This permits a rise in CO levels in the vicinity of 400°C at the transition between thermal regimes, as seen in Fig. 10, and an increase in methane concentrations for biomass feedstocks at about 450°C, as observed in Fig. 12. Cellulose pyrolysis reactions during the interval between 350°C and 450°C presented by Kawamoto et al. (2003) involve the decomposition of levoglucosan to low-molecular-weight volatile products or polymerization to polysaccharides releasing CO and CO2 and forming a nonreactive carbonized layer.
CH4 levels remain low throughout the gasification process, by at least an order of magnitude, for all of the fuels currently studied. During steam gasification, as the temperature rises, the H2 and CO concentrations increase. Jangsawang et al. (2006a, 2006b) noticed that the H2 and CO concentrations resulting from the steam gasification of cellulose remained low below 540°C. A marked increase in H2 levels occurred above 540°C, with a limiting temperature of about 930°C after which the H2 and CO concentrations began to decline. Jangsawang et al. (2006b) attributed the drop in H2 production as being due to competing processes that result in H2 destruction due to molecular dissociation reactions, which become significant at about 1,000°C, and H2 formation due to the decomposition of the biomass during steam gasification. Only for no CO2 co-feed was there a consistent black char residue present for the biomass samples. This high-temperature biomass char residue was available to enter into the high-temperature steam gasification reactions, whereas with only 10% CO2 injection nearly all of the biomass was exhausted before 950°C. Many of the rapid drops in gas concentrations, such as that appearing in Fig. 10 for the CO evolution, were attributable to the exhaustion of the biomass sample at the high gasification temperatures, particularly for the higher CO2 levels.
Following low-temperature dehydration that occurs before 100°C, degradation of the lignocellulosic structural components can be characterized by a set of coupled mechanisms involving both radical intermediates and molecular species. Mineral impurities in the biomass exert a catalytic effect that can influence both reaction rates and product selectivity. Free radical reactions govern and sustain the thermal treatment in the temperature interval between 500°C and 1,000°C. Initiation reactions become significant above 200°C and this corresponds to the transition temperature at which mass loss becomes significant for all of the carbon-based fuels that were studied. During gasification of the various feedstocks, the steam and CO2 can access the carbon structure through both low- and high-temperature reactions resulting in a condensed structure that is more thoroughly processed to volatiles at high temperature through the use of CO2. Biomass decomposition results in low-temperature fragments that have short carbon chains or 5–6 carbon ring structures and a variety of oxygenated species characteristic of the polysaccharide cellulose/hemicellulose and highly crosslinked aromatic phenylpropanoid lignin composition from which they originated. Characteristic of hydrocarbon thermal treatment whether through gasification or combustion processes, H abstraction and β-carbon scission reactions play a significant role in the degradation process. Radical reactions continuously feed into the coupled mechanisms. Initiation and chain propagation reactions that involve the cleavage of branched OH radicals from the cellulose chain as well as CH3 radicals from the methoxy-phenylpropanoid lignin structure help to sustain the thermal treatment.
At high steam gasification temperatures above 700°C, levels of CO rise because of the Boudouard reaction in which CO2 available in the flow reacts with residual carbon fuel. With increasing levels of CO2 introduced into the flow, a strong CO evolution enhancement occurs, which can be seen in Fig. 10 for beachgrass II (steam-to-carbon, S/C = 18). As the pyrolysis char continues to be heated, residual carbonyl groups are decarboxylated to CO and CO2. A mechanism proposed by Banyasz et al. (2001) for the slow heating rate low-temperature pyrolysis of cellulose involves production of char, tar, CO2, H2O, and low-molecular-weight volatiles (mainly aldehydes) that are responsible for biomass-derived CO evolution at low temperatures in the absence of CO2 injection. During pyrolysis, any available oxygen either brought into the gasifier as H2O or CO2 influent, present as CO, or evolving from the biomass structure itself can be adsorbed by the highly reactive char. With subsequent heating, this char-adsorbed CO is released, providing a continuous supply of CO that can account for the uniformly increasing concentrations of CO above 500°C, as seen in Fig. 10. The lignin decomposition mechanisms developed by Demirbas (2006) and Kawamoto et al. (2003) involve CO evolution and carbonization reactions that occur at about 400°C and can help to account for the low-temperature production of CO. The CO evolution signal was the strongest and most consistent of those observed. CO enhancement with increasing CO2 injection was observed for all woods and grasses studied.
The steam-reforming reaction becomes significant at temperatures above 550°C because of high concentrations of CO2 and H2O relative to CO and H2. This gasification reaction is responsible for the abrupt increase in H2 production that occurs at about 600°C seen for all CO2 injection ratios in Fig. 11 for poplar (S/C = 21). H2 evolution depression is clearly visible with increasing percent of CO2 injection. This strong depression of the H2 mole fraction was only observed for those feedstocks having lower S/C ratios. For a given influent steam rate, this lower S/C value represents a higher carbon input rate. Lower S/C samples have the potential for higher carbon conversion and a more pronounced effect because the gasification process was run with excess H2O and CO2 to ensure that the carbon feedstock was not the limiting factor.
The steam-reforming, Boudouard, and RWGS reactions are high-temperature reactions common to all hydrocarbon gasification processes. The Boudouard and RWGS reactions along with desorption of CO from the char drive the reverse steam gasification reaction, resulting in a decrease in H2 evolution concentrations with increasing CO2 injection rate that should become more pronounced above 700°C. This H2 depression was observed in many of the carbon feedstocks, including grass, wood, and residues, particularly at the low S/C ratios. A biomass decomposition mechanism proposed by Demirbas involves H abstraction from either a donor molecule or a high-molecular-weight fraction that provides the H necessary to stabilize a biomass free radical. Biomass having a higher cellulose content undergoes degradation earlier and so will stop consuming these stabilization radicals earlier and result in a H2 evolution rise earlier than high lignin content biomass. We experimentally verified this, because the grasses, lower in lignin content, were consistently observed to show H2 evolution onset earlier than the woods, higher in lignin content, and so slower in mass decomposition and H2 evolution onset.
Although methane evolution levels remain consistently low throughout the gasification process, when the woods and grasses are undergoing a cluster of radical reactions cleaving off the methyl and methoxy radicals from the lignin structure between 300°C and 450°C, they result in low levels of methane during pyrolysis and low-temperature gasification. These direct hydrogenation reactions result in a continual supply of methane. At the transition between regimes, at about 400°C, the H2 and CO levels begin to rise resulting in a corresponding increase in the CH4 evolution as seen in Fig. 12 for beachgrass I (S/C = 5.5). As a consequence of the H2 depression due to CO2 injection, a corresponding CH4 depression commencing during the same temperature interval can be observed in Fig. 12. This CH4 depression results from the decreased H2 levels available to enter into the methanation reactions.
What we found, concerning the isolation of the two structural components, is shown in Fig. 13. The decomposition profiles shift to lower temperatures at a slower heating rate. At the slowest rate in this study (1°C min−1) and using CO2 as the gasification medium, the cellulose decomposed at the lowest temperature leaving the highest residual ∼75% of the lignin to be subsequently processed whether by thermal or chemical means. At the highest heating rate studied (100°C min−1) less differential isolation was possible with only 65% of the lignin remaining following degradation of the cellulose that was completed at a later temperature of 450°C during the faster heating rate program. Slower heating rate during CO2 gasification offers a selective advantage when lignin isolation and conversion is the goal of the biomass treatment process. It enables processing of the cellulosic component within a narrow temperature window during which the decomposition rate of the thermally resistant lignin permits a greater fraction of the lignin component to survive the low-temperature pyrolysis treatment. The ability to perform the thermal treatment at a lower temperature, using CO2 as a valuable processing medium, without the cost and energy consumption necessary to heat the water used during steam gasification offers a distinct advantage. As adequate supplies of industrial and commercial water become more scarce, there will be a shift toward less water-intensive processes. The distinction between the thermal processing in steam versus CO2 can be seen in Figs. 13 and 14. Whereas the slow 1°C min−1 heating rate for steam gasification left nearly 40% of the lignin still unprocessed to volatiles by 930°C, the slow heating of the lignin and cellulose components in a CO2 gasification medium resulted in complete conversion by 930°C, with only a 3% ash/mineral residue remaining from the lignin and only 0.4% remaining from the cellulose. The mass decomposition curves for two representative biomass feedstocks processed under steam (0%) and CO2 (100%) gasification are shown in Fig. 15. Blue fir needles, higher in lignin content, begins pyrolytic degradation earlier (200°C) and has a slower rate of decay. Poplar wood, higher in cellulose content, begins pyrolytic degradation at a later temperature (250°C) and shows a much faster rate of decomposition during pyrolysis. The greatest difference between H2O and CO2 gasification is seen after 900°C, where rather than leaving 22% of the needles unprocessed, less than 2% ash remains using CO2. Similarly, rather than 12% of the wood remaining unprocessed, less than 1% ash results using CO2.
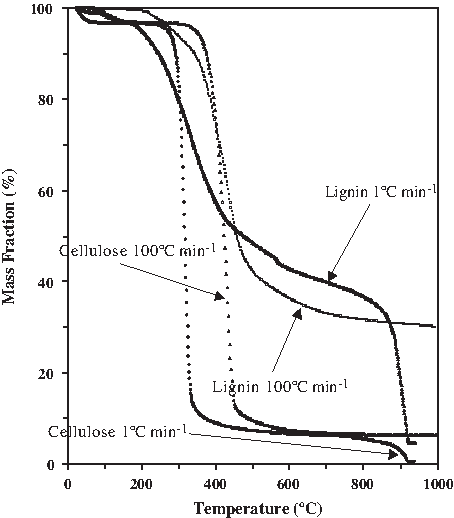
Mass decomposition curve comparing lignin and cellulose, 100% CO2, at 1°C min−1 and 100°C min−1.
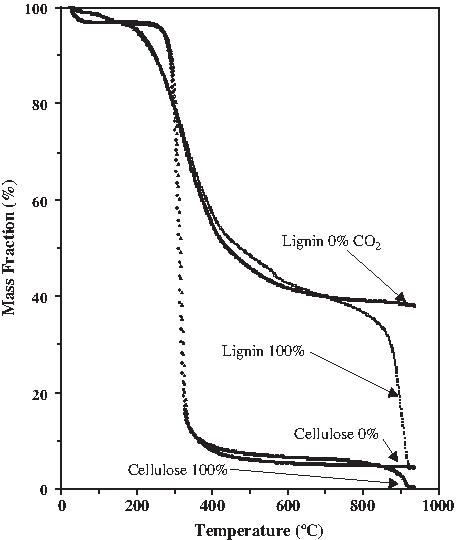
Mass decomposition curve for lignin and cellulose, 1°C min−1, comparing steam (0%) and CO2 (100%) gasification.
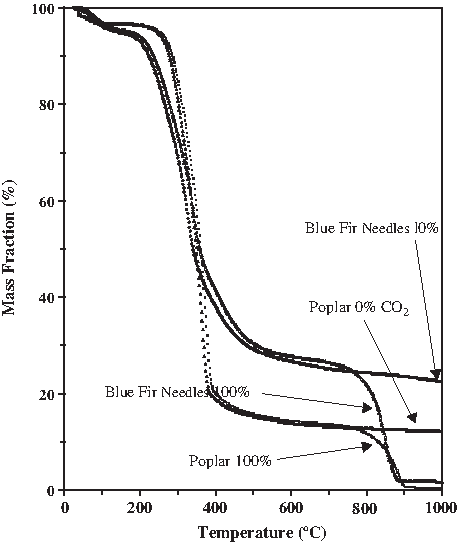
Mass decomposition curve for two representative biomass feedstocks, 10°C min−1, comparing steam (0%) and CO2 (100%) gasification.
Organosolv lignin was chosen as the lignin model because it was deemed most likely, of the industrial lignins that were available, to still retain a significant proportion of those structural units and type of bonding found in naturally occurring lignin. Its relatively mild preparation minimized the breaking of side chains and significant cleavage of ether bonds in the phenolic structure commonly found in lignins prepared by the Kraft (alkali) process. Organosolv lignins are typically derived from hardwoods because the pulping process, in aqueous alcohol with mineral acids, is more efficient with hardwoods than softwoods. Although Organosolv lignin is very similar to alkali lignin, the higher level of depolymerization and the amount of sulfur present in Kraft lignins, because of processing with NaOH and Na2S, is not present in the milder Organosolv lignin that is sulfur free (Sigma Aldrich Chemicals, 1998). Characteristic of Kraft pulping is the hydrolysis of alkyl–aryl–ether bonds that can result in significant departure in behavior from high-lignin content biomass feedstocks by a Kraft lignin surrogate model. Hydrolytic lignin also involves a more extreme processing of a typically herbaceous feedstock such as bagasse (the high-lignin structural component of sugarcane stalks). The biomass source is superheated with steam using an acetic acid catalyst where isolation is finally accomplished in an NaOH solution. We chose Organosolv lignin as the reagent most similar to the natural lignin occurring in the collection of actual biomass feedstocks because it underwent what appeared to be the most benign and least chemically intrusive preparation: isolated by membrane filtration of the spent pulping liquor from hardwood chips dissolved in a mineral acid aqueous ethanol solution.
Natural unprocessed lignin was found by Saiz-Jimenez and de Leeuw (1984) to have a higher degree of crosslinking and more intact side chain branches than the industrial lignin. Alkali lignin was also found to yield a greater portion of oxidized species such as guaiacol and vanillin derivatives. In contrast to the softwood spruce whose pyrolysates contained mostly guaiacyl units (one methoxy), the milled wood lignin from the hardwood beech studied by Genuit and Boon (1987) contained comparable amounts of both guaiacyl and syringyl units (two methoxy groups), indicating a higher level of methoxylation. Depending on the pyrolytic depolymerization mechanism, this would have a significant impact on methane evolution and the methylation products in the vicinity of 400°C following low-temperature pyrolysis. Different CH4 evolution profiles were observed for grasses, having lower lignin/cellulose ratios and higher phenol fractions (with no methoxy groups) that have been correlated with the herbaceous-specific monolignol p-coumaryl. The relatively high level of phenol derivatives in the lignin degradation products was observed by Faix et al. (1987) in the milled wood lignin of bamboo. They identified and quantified 75 phenolic pyrolysates and observed a significant variation in the product mole fractions between feedstocks (beech, spruce, bamboo, teak).
Our next set of experiments will involve the detection and characterization of pyrolytic degradation products at slow heating rate for the various biomass feedstocks under an inert medium, and then these profiles will be compared with those obtained using H2O and CO2 gasification environments to contrast the pyrolysate evolution profiles as a function of temperature using a constant heating rate program for the TGA. The GC/mass spectrometry (MS) analysis will enable identification of the 30 significant lignitic thermal degradation species that we have selected for study. We have found that differences in number and type of structural units present in either a natural biomass lignin (wood, grass, or residue) or a surrogate synthetic molecule (e.g., Organosolv lignin) as well as the gasification medium (H2O/N2, CO2) and the heating rate (1–100°C min−1) result in different gas evolution profiles for CO, CH4, H2, and CO2. Organosolv lignin pyrolysate behavior can be expected to deviate from natural milled wood lignin because of the pulping process. As a result, a significant portion of the alkyl–aryl–ether bonds are broken, resulting in a relatively higher amount of phenolic hydroxyl groups than that found in the natural biomass source. Though the surrogate models offer more detailed information regarding individual component decomposition products that gives insight into likely mechanistic pathways, ultimately the actual feedstock needs to be tested. Through this type of fundamental research, a comprehensive database characterizing the chemical feedstock potential and nature of pyrolytic degradation products can be compiled under varying gasification conditions for the wide array of promising biomass feedstocks whose conversion is commercially, socially, and environmentally viable.
Pulverized wood samples were prepared from untreated planks of wood. Grasses were prepared by grinding and ball milling with extreme care so as not to mechanically work the sample to a point where it could experience sufficient heating and subsequent thermal degradation. Sand grains were carefully removed from the beachgrass, but no washing of the grasses was done because mineral leaching could occur in the rinse water. The bark and all of the herbaceous materials, including the green undegraded pine and blue noble fir needles, were dried and then ball milled. The cellulose model chosen, for comparison with the biomass structural component, was a 20 μm microcrystalline powder from Sigma Aldrich Chemicals selected for its purity, particle size, moderate moisture content (< 7%), and low heavy metal content (Sigma Aldrich Chemicals, 2003).
We have extracted representative kinetic parameters to assist in modeling efforts as well as classify the reaction orders for the feedstock tested. A least squares minimization of the rate of weight loss fraction enabled extraction of global decomposition kinetic parameters. As evidenced in Table 3, it is clear that lignin is of higher reaction order than cellulose. We have determined lignin as having reaction order near 3 and cellulose having a value closer to 1.
The actual feedstocks had reaction orders intermediate between these values, roughly corresponding to the relative cellulose versus lignin content. For example, those with reaction order 1 typically have higher cellulose content, whereas those whose global decomposition reaction order is closer to 2 had, in general, a higher lignin to cellulose content. During CO2 pyrolysis, at a heating rate of 5°C min−1, typical activation energies were about 49 kJ mol−1 for lignin and 219 kJ mol−1 for cellulose. The slow thermal processing of lignin over a broad temperature range from 100°C to 800°C is a result of the greater thermal resistance of the lignin structure during pyrolytic decomposition below 500°C, when compared with cellulose whose processing is nearly complete before low-temperature gasification. This is evidenced in the higher reaction order of 3 for lignin when compared with 1 for cellulose during pyrolysis.
Finally, we have developed a ballistic heating rate procedure that can be used for fast pyrolysis investigation. During the rapid heating, from 25°C to 1,000°C in less than 2 min time, we have observed the evolution of gaseous chemical species ranging from hydrogen to propane. While typical TGA studies employ constant heating rates of ∼1–50°C min−1, the uncontrolled rapid pyrolysis heating rate varied from 40°C s−1 to a leveling off of 10°C s−1 at the completion of the mass decomposition near 1,000°C. To achieve ballistic heating, the Instrument Specialists Temperature Programmer Interface was coupled to the Dupont 951 TGA to act as the Thermal Analyzer. It sets the temperature program by means of a PID controller. By manually nulling out the PID in the software, the furnace can control the temperature felt by the quartz tube, enabling the sample thermocouple to be removed from the loop, with ballistic heating capabilities once the quartz tube is injected into the preheated furnace. Figure 16 shows the evolution of low-concentration species as a function of temperature during the ballistic heating of commercial pulverized wood shavings.
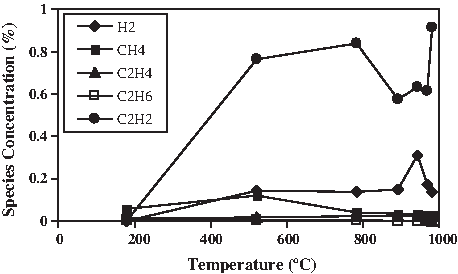
Gas evolution of minor species, from pulverized wood shavings during ballistic heating in air at 1,000°C.
O2 brought in by the air resulted in lower levels of H2 than were observed under inert environments of N2 and Ar. Although H2 levels rose during gasification under ballistic heating conditions in air, the drop above 950°C was attributed to exhaustion of the biomass. The levels of H2 observed during wood gasification in H2O/N2 at 10°C min−1 were of the same order of magnitude as those observed in air at ballistic heating rates (∼30°C/s), at about 0.2%. At 1,000°C following processing in air, nearly all that remained in the pan was a mineral ash residue. Acetylene (C2H2) levels during ballistic heating remained high and nearly constant at about 0.7%, much higher than any levels detected during slow TGA thermal treatment at 10°C min−1. The pulverized wood processed in air had a corresponding mass loss from 98% to 11% as the temperature rose from 129°C to 619°C during an interval of 13 s. Much of the biomass devolatilization occurred in this interval. Peak CH4 levels observed for woods and grasses processed in H2O/N2 at 10°C min−1 were slightly higher at 0.15–0.25% when compared with 0.1–0.15% for the pulverized wood under ballistic conditions in air. The actual difference in quantity of CH4 evolved is significant because the interval over which peak CH4 evolution occurs corresponds to that of maximum weight loss. Those pathways enabling cleavage, depolymerization, condensation, and recombination reactions of the lignocellulosic structure during slow pyrolysis that result in the onset of CH4 evolution at much lower temperatures, ∼250°C, as well as the absence of measurable levels of C2–C4 hydrocarbons, suggest that different thermal degradation mechanisms are in operation under ballistic conditions. This would indicate that residence time and transport limitations result in a quantifiable difference in the distribution of pyrolysates. Additional evidence was the presence of condensable volatiles (bio-oils) on the cooler portions of the reactor tube furnace that were identified by the GC/MS as oxygenated species.
Simultaneous measurements were made for high-concentration species as shown in Fig. 17. The results show that during rapid heating, reactions begin at low pyrolytic temperatures and continue throughout thermal treatment into the high temperature gasification range until all of the feedstock is consumed. Significant levels of viscous pyrolytic tar were observed to condense onto the cooler external surfaces of the quartz furnace during ballistic heating, although no such liquid deposition was observed during slow TGA processing. Subsequent GC/MS analysis identified many of these species as aldehydes, ketones, and phenolic derivatives.

Gas evolution of major species, from pulverized wood shavings during ballistic heating in air at 1,000°C.
Conclusions
An experimental kinetic investigation was conducted, which examined the decomposition products resulting from the CO2-enhanced gasification of a variety of biomass feedstocks that included grasses, woods, and agricultural and forestry residues as well as model cellulose and lignin compounds. Two corresponding thermal regimes, representing both mass decay and gas evolution profiles, exhibited two corresponding thermal regimes, whose transition was near 400°C, for all of the lignocellulosic feedstocks studied. At a heating rate of 10°C min−1, more complete processing of the biomass samples were possible when undergoing CO2 when compared with steam gasification. Whereas steam gasification resulted in a large black char residue, use of 30% CO2 to enhance the thermal processing of walnut shells resulted in a small light mineral residue, with less waste for postprocessing following thermal treatment. Use of both H2O and CO2 introduced into the gasification medium results in more reactive char, although CO2 was observed to create a more porous char structure with increased surface activity. The enhanced pore structure and the ability of CO2 to access this elaborate network of channels resulted in a more thorough processing of the biomass feedstocks. Though the polysaccharide cellulosic structural component of biomass undergoes thermal decomposition at lower pyrolytic temperatures, <450°C, the aromatic and highly crosslinked thermally resistant lignin component is still undergoing degradation >750°C. A distinctly different char structure was observed for lignin undergoing CO2 rather than H2O gasification. Lignin that underwent steam gasification exhibited a hard low porosity surface coating that resulted in less surface area exposed to the H2O gasifying medium, and consequently, a lower level of conversion during steam gasification at high temperatures. During CO2 gasification of lignin, a highly porous char structure developed, which offered greater surface area and access through an intricate network of channels. This enables the CO2 to penetrate the inner volume of the lignin char, resulting in more thorough processing. Although during high-temperature processing it is the reactive gasification medium that plays a significant role in the more complete conversion to volatiles, at lower pyrolysis temperatures it is the heating rate that is most significant in enabling thermal decomposition and selective separation of lignocellulosic components. Comparing the mass decomposition curves for various biomass feedstocks during CO2 gasification, the rates were found to be intermediate between those of lignin and cellulose, although the decay began earlier for the herbaceous feedstocks such as beachgrass, alfalfa, and blue fir needles, when compared with the model lignocellulosic structural surrogates. Grasses and agricultural and forestry residues were observed in the SEM/EDX analysis to have higher alkaline levels, indicating that actual feedstocks may be exhibiting a catalytic effect not present during the decomposition of the pure structural components. The relative amounts of the lignocellulosic components surviving thermal treatment can be altered by adjusting the CO2 level in the gasification medium as well as the heating rate of the furnace. While the cellulosic component can be processed at low temperature, the remaining lignin, which is more thermally resistant and can survive pyrolytic degradation, can undergo subsequent processing.
The largest percent mass loss in the biomass samples occurred during pyrolysis between 250°C and 400°C at which time the structural components were undergoing depolymerization and condensation reactions. In a CO2 medium at both slow (1°C min−1) and fast (100°C min−1) TGA heating rates, mass decomposition levels greater than 80% of the cellulose and 45% of the lignin were observed. Mass decay profiles for the Organosolv lignin began earlier than the powdered cellulose, corresponding to a lower activation energy calculated for CO2 lignin pyrolysis, 49.3, when compared with cellulose pyrolysis having a much higher value of 219.0 kJ mol−1. Also, the thermal decomposition of lignin occurred over a much wider temperature interval through the high-temperature gasification range as indicated by a higher global reaction order of 3 for lignin when compared with 1 for cellulose.
The most significant difference in biomass processing in CO2, when compared with steam gasification, occurred above 750°C where nearly all of the biomass was converted to volatiles with less than 2% ash remaining after CO2 gasification when compared with 12% cellulose and 22% lignin during steam gasification. Although the low-temperature mass decay pyrolysis profiles were nearly independent of percent CO2 introduced into the gasification stream, they were highly dependent upon heating rate. The high-temperature gasification profiles were distinctly different between CO2 and steam gasification because only when pure CO2 was used as the gasification medium under a slow heating rate did complete processing of the components to volatile products occur.
CO evolution for the biomass feedstocks begins after 600°C, with enhanced CO production corresponding to increased levels of CO2 introduced that can enter into the Boudouard reaction at high gasification temperatures. A local maxima in CO concentration occurring in the vicinity of 400°C can be attributed to lignin decomposition involving CO evolution and carbonization reactions. The three principal reactions governing thermal degradation at high gasification temperatures are the Boudouard, RWGS, and steam-reforming reactions. The net effect of injecting higher levels of CO2 and lower levels of H2O into the gasifier is to favor the production of increased levels of CO and decreased levels of H2 during high-temperature gasification. The ability of CO2 to create a distinctly different micro- and macroporous char structure enabled a 30% CO2 injection to result in a small light mineral residue compared with a larger black char volume without CO2 during walnut shell steam gasification. The Boudouard reaction between the CO2 and the macropore carbon skeleton occurring at elevated gasification temperatures is the most significant reaction responsible for the high-temperature slow heating rate complete processing of the feedstock.
H2 evolution for the biomass fuels also begins after 600°C and is enhanced because of addition of H2O that enters into the steam-reforming reaction at high temperatures. The Boudouard and RWGS reactions along with desorption of CO from the char drive the reverse steam gasification reaction resulting in a decrease in H2 evolution concentrations with increasing CO2 injection rate that becomes more pronounced during gasification above 700°C. The onset of H2 evolution may also be attributable to the cessation of H abstraction reactions that are continually consuming H to help stabilize the biomass free radicals during thermal treatment. These H radicals become available to form molecular H2. Cellulose degradation begins slightly later than lignin decomposition, in agreement with a significantly higher calculated global activation energy for cellulose when compared with lignin, but cellulose decomposition is completed at a lower temperature. Those biomass feedstocks higher in cellulose content and lower in lignin content would tend to exhibit an earlier thermal decomposition and incipient H2 evolution corresponding to the cessation of H consumption for biomass radical stabilization. This earlier onset was experimentally observed for grasses, whose lignin content is lower than that of woods.
Whereas steam was observed to significantly improve the H2 production at elevated temperatures, CO2 was observed to enable a more complete processing of the lignin component at higher temperatures. Once low-temperature pyrolysis is complete at ∼375°C, the cellulose component has undergone nearly all of its conversion to volatiles but a significant portion, ∼70%, of the lignin remains. By varying the heating rate of the TGA furnace as well as the %CO2 introduced into the gasification medium, optimum conditions under which separation of the greatest portion of lignin from the cellulosic biomass components occurred were identified. Isolation of the two structural components was observed during slow thermal treatment using CO2 with thermal processing of the cellulosic component at low temperature and the thermally resistant lignin residual undergoing thermal degradation at higher gasification temperatures. The decomposition profiles shift to lower temperatures at a slower heating rate. At the slowest rate in this study (1°C min−1) and using CO2 as the gasification medium, the cellulose decomposed at the lowest temperature, leaving the highest residual ∼75% of the lignin to be subsequently processed. At the highest heating rate (100°C min−1), less differential isolation was possible, with only 65% of the lignin remaining following degradation of the cellulose that was completed at a later temperature of 450°C during the faster heating rate program. Slower heating rate during CO2 gasification offers a selective advantage when lignin isolation and conversion is the goal of the biomass treatment process. It enables processing of the cellulosic component within a narrow temperature window during which the slower decomposition rate of the thermally resistant lignin permits a greater fraction of the lignin component to survive the low-temperature pyrolysis treatment. The ability to perform the thermal treatment at a lower temperature, using CO2 as a valuable processing medium, without the cost and energy consumption necessary to heat the water used during steam gasification offers a distinct advantage.
The kinetic analysis yielded global decomposition parameters useful for modeling as well as gaining some insight into the similarities between CO2 and H2O/N2 pyrolysis kinetics. Gasification kinetics is significantly different between the two media and a study is currently underway to compare gasification parameters. Only slow (<10°C min−1) heating rates and small sample sizes (<25 mg) can accurately represent the global decomposition parameters to avoid heat and mass transfer limitations that significantly alter the derived kinetic parameters. Lignin was determined to be best represented by a third-order global reaction order, whereas that of cellulose was closer to first order. The actual feedstocks had reaction orders intermediate between these values (1–2), roughly corresponding to the relative cellulose versus lignin content. During CO2 pyrolysis, at a heating rate of 5°C min−1, typical activation energies were about 49 kJ mol−1 for lignin and 219 kJ mol−1 for cellulose. The slow thermal processing of lignin over a broad temperature range from 100°C to 800°C is a result of the greater thermal resistance of the lignin structure during pyrolytic decomposition below 500°C, when compared with cellulose whose processing is nearly complete before low-temperature gasification. This is evidenced in the higher reaction order of 3 for lignin when compared with 1 for cellulose during pyrolysis.
During ballistic heating of wood shavings, from 25°C to 1,000°C in ∼1.5 min, evolution of gaseous chemical species ranging from hydrogen to butane was observed. As a result of rapid heating, volatile evolution from decomposition reactions begins at low temperature and continues throughout thermal treatment into the high temperature range until all of the feedstock is consumed. Significant levels of viscous pyrolytic tar were observed to condense onto the cooler external surfaces of the quartz furnace during ballistic heating, although no such liquid deposition was observed during slow TGA processing. Subsequent GC/MS analysis identified many of these species as aldehydes, ketones, and phenolic derivatives. While slow TGA heating to pyrolytic temperatures was conducive to solid char formation and heating to gasification temperatures to volatiles formation, ballistic heating was conducive to both gaseous and condensable volatiles (liquid tar) formation.
Footnotes
Author Disclosure Statement
No competing financial interests exist.