
Abstract
Abstract
Thiosulfate (S2O32−) hosts redox stability in time frame of days, rendering it a candidate ion exchange competitor in anoxic groundwaters during the brief time frames of treatment processes. Indeed, this research determined that the oxyanion thiosulfate (S2O32−) competed with perchlorate (ClO4−) for sorption onto activated carbons that had been preloaded with a quaternary ammonium surfactant. Moreover, this research showed that when proper prechlorination oxidized this S2O32− to sulfate (SO42−), this competition was diminished. When rapid small-scale column test employed Redlands, CA, groundwater that contained a native 30 μg/L ClO4−, this exhibited a 6 μg/L ClO4− breakthrough after 33,000 bed volumes (BVs) when processed through bituminous-based granular-activated carbon that had been preloaded with a quaternary ammonium surfactant, namely, 0.24 g/g dicocoalkyldimethylammonium chloride (Arquad 2C-75). When this same water was spiked with 1,000 μg/L S2O32−, 6 μg/L of ClO4− broke through at 17,000 BV. However, when S2O32− spiked Redlands groundwater was also spiked with 2,500 μg/L chlorine, this reactant stoichiometrically oxidized the S2O32− so as to diminish this competition, such that 6 μg/L ClO4− broke through at 31,000 BV. Similar rapid small-scale column test trends were exhibited when using deionized distilled water that had been spiked with ClO4−, S2O32−, and chlorine.
Introduction
Sulfur redox species
Sulfur intermediates can persist for times measured in seconds to decades, as dictated by the environmental conditions that are present. For example, S2O32− is stable for days at pH conditions that are near neutral. This constitutes significant durations relative to the time that water resides within a water treatment facility. Among the sulfur intermediates, S2O32− is one that can persist for times measured in days when in the presence of incidental air contact and at a near-neutral pH (Rolla and Chakrabarti, 1982; Millano et al., 1983; Zhang and Dreisinger, 2002; Breuer and Jeffrey, 2003a, 2003b; Lam and Dreisinger, 2003; Senanayake, 2004).
Thus, thiosulfate can persist for days in groundwaters that are pumped to the surface. Thiosulfate, as an oxyanion, could compete with the oxyanion perchlorate on an ion exchange-type medium. Indeed, in the ion chromatography analysis (which is based on similar ion exchange functional sites—see below), the thiosulfate ion peak occurs in the same time span as does perchlorate. Despite this prospect, the authors are not aware of any other refereed journal articles that have appraised the competition of S2O32− with ClO4−, for exchange sites of quaternary ammonium functional groups. The work herein aims to bridge this void.
Motivation for hypothesis regarding intermediate sulfur oxyanions
Our Penn State team has developed a water treatment technology where granular-activated carbon (GAC) has been tailored with quaternary ammonium surfactants or ammonia thermal treatments to increase the bed life of the carbon for ion-exchanging ClO4− and other oxyanions (Chen and Cannon, 2005; Chen et al., 2005a, 2005b; Parette and Cannon, 2005; Parette et al., 2005). The surfactant-tailored GAC increased the carbon bed life 33 times relative to conventional GAC, when processing water with 70 μg/L ClO4− from Redlands groundwater to below 6 μg/L via rapid small-scale column tests (RSSCTs) (Parette and Cannon, 2005; Parette et al., 2005). This 6 μg/L is the perchlorate action level in California regulations. Demonstration and pilot scale studies that were conducted at the Texas Street Water Treatment Plant depicted ClO4− breakthrough in fewer bed volumes (BVs) than when conducting RSSCT trials that employed barrels of the Redlands water that had been stored in Penn State for a month or longer. However, when RSSCTs employed Redlands groundwater that was employed as soon as it reached Penn State, ClO4− breakthrough occurred considerably sooner than when employing this same groundwater after it had been stored for a month. RSSCTs with fresh barrels of Redlands water exhibited breakthrough at about the same time as for our full-scale demonstrations. This led our team to suspect that a competing oxyanion intermediate that gradually oxidizes was adversely impacting perchlorate adsorption. This was further corroborated by the negative to low redox values that were monitored at the Texas street treatment plant, but positive redox levels monitored in the groundwater that had been stored in barrels for several months (Patterson, 2009). The validity of redox probe measurements have been questioned by some scientists. This raised the intriguing hypothesis that the Redlands groundwater contained a species that in its reduced or intermediate-valence form competed quite significantly with ClO4−, but in its oxidized-valence form did not compete as prominently.
Objective and hypothesis
In light of this background, our specific objective was to test the hypothesis that S2O32− competitively diminished ClO4− sorption on GAC that had been preloaded with quaternary ammonium surfactant. Moreover, we hypothesized that stoichiometrically designed chlorination dosing could oxidize S2O32− to SO42−, and then the SO42− would not extensively diminish the bedlife for ClO4− ion exchange.
Chlorine oxidation of intermediate-valence sulfur oxyanions
This article has appraised chlorine as an oxidant for converting S2O32− to SO42− so that the sulfur species incurs less sorption competition for ClO4−. The rationale for this has been that chlorine is already included at the Redlands (and other) well-head treatment sites, and parallel tests showed that the sulfate competed considerably less than thiosulfate for exchange sites (see below). Thiosulfate is also routinely used as a dechlorinating agent in analytical labs (Tchobanoglous et al., 2003). Stoichiometrically, a molar ratio of 4 mol of chlorine (and thus 4 mol of hypochlorous acid) to 1 mol of S2O32− will result in complete oxidation of S2O32− to SO42− per Equation 1:
Chlorination treatment processes also influence activated carbon since the activated carbon serves as a redox surface that can reduce chlorine to chloride while the carbon surface becomes oxidized (Hassler, 1967; Snoeyink et al., 1981; Hwang et al., 1989; Martin and Shackleton, 1990; Dixon and Lee, 1991; Gonce and Voudrias, 1994; Collivignarelli et al., 2006). Indeed, as an unfavorable side effect incurred via excessive chlorination, the activated carbon surface can experience oxidative degradation, yielding oxygenated substituents (Snoeyink et al., 1981) such as carboxyls whose negative charge could repel the ClO4− anion. Also, under the conditions herein, there was a possibility that chlorine could oxidize the reduced –N in the quaternary ammonium functionality. If this occurred it could inactivate the quaternary ammonium's ability to exchange with perchlorate. However, tests showed that such ammonium oxidation did not occur for the conditions herein, as described below.
Experimental Protocols
Materials
The GAC that was used in these experiments was bituminous-based UltraCarb unless otherwise identified. This activated carbon was obtained from Siemens Water Technologies (formerly USFilter). One experiment employed another bituminous-based activated carbon from Superior Adsorbents Inc. These carbons were ground and sieved to a mesh size of US # 200 × 400, which was 38 × 75 μm.
Unless otherwise listed, the groundwater for these experiments originated Redlands, CA, Texas Street Well 31A. This native groundwater contained 30 μg/L ClO4−, 55 mg/L SO42−, and 38 mg/L nitrate as NO3− (Table 1). When fresh, this Redlands groundwater also contained some intermediate-valence sulfur species, as discussed below. The Redlands water was stored in 55-gallon polyethylene barrels at ambient temperature, and when not in use, they were sealed with polyethylene caps that were not airtight.
Some tests employed earlier-sampled Redlands groundwater that contained 52 μg/L ClO4− and 10 mg/L nitrate as NO3−.
Several initial experiments employed Redlands groundwater that was sampled earlier (identified as earlier-sampled herein), and this contained 52 μg/L ClO4− and 10 mg/L nitrate as NO3−.
The cationic surfactant used was dicocoalkyldimethylammonium chloride (Arquad 2C-75), unless otherwise listed. Arquad 2C-75, with an active molecular weight of 397 Da, was chosen among the available quaternary ammonium surfactants because it has two long carbon chains that facilitate low leaching of the surfactant from the carbon (Parette and Cannon, 2005; Parette et al., 2005). Arquad 2C-75 were prepared from analytical-grade reagents that were dissolved in deionized distilled water. Several experiments used tallowalkyltrimethylammonium chloride (Arquad T-50) or cetylpyridinium chloride. Previous experiments had shown that the preloading of these three surfactants offered about the same bed life for perchlorate removal (Parette and Cannon, 2005).
Methods
Preparation of sodium thiosulfate solution and other solutions
All solutions were prepared from analytical-grade reagents and deionized distilled water, unless otherwise listed. The S2O32− source was sodium thiosulfate by Fisher Scientific. Thiosulfate spiking concentrations and analysis standards were prepared from a 1 g/L as S2O32− stock solution. During RSSCT experiments, thiosulfate stock solutions and influent solutions were prepared every 3 days in a given RSSCT run period. This ensured that a relatively uniform S2O32− concentration was used at all times (as discussed below). To test the rate of S2O32− degradation with incidental air contact, 1 mg/L of S2O32− was mixed into 200 mL of deionized distilled or Redlands water and let stand without further agitation in capped bottles. Thiosulfate was tested at T = 0, 1, 2, 3, 4, and 5 days. The same protocol was also used to monitor the rate of 1 mg/L sulfide degradation in incidental air contact.
Rapid small scale column tests
The RSSCTs were designed for proportional diffusivity according to Parette and Cannon (2005). The RSSCT columns were dry- packed with UltraCarb, and glass wool was packed at both ends. About 1.27 g of UltraCarb GAC was used and tailored with 33 BVs of 0.4% Arquad 2C-75 solution that was recirculated through the bed for 3 days, during which the surfactant solution was pumped and recirculated at a rate of 2 mL per minute through the carbon medium. The surfactant-tailored GAC was not washed before the introduction of the ClO4−-contaminated influent water solution. This protocol had been observed to preload 0.24 g Arquad 2C-75/g GAC, which correspond to 0.56 meq of active N functionality/g GAC (Parette et al., 2005).
The RSSCTs were conducted at ambient temperatures, which were measured at 20°C ± 2°C, and the flow rate was maintained at 2.5 mL/min through a 2.65 mL bed, giving a 1.1 min empty bed contact time (EBCT) through the RSSCT. Per proportional diffusivity, this would simulate an EBCT of 10 min for the US # 12 × 40 mesh size GAC (1.7 × 0.425 mm) that could be used at the field scale level. This would also simulate a 5.4 min EBCT for US # 20 × 50 mesh size GAC (0.85 × 0.30 mm). Pumps (Waters models 501 and 510) were used to provide flow.
Chlorine gas preparation and application
Hypochlorous acid was produced by reacting potassium permanganate with HCl, then bubbling the product chlorine gas into a plastic polyethylene collapsible container that contained deionized distilled water (Patterson, 2009). The free chlorine stock solution concentration was monitored using the HACH DR 2800 spectrophotometer; while the HACH CN-66 color wheel model was used to measure sample concentrations daily to ensure that the correct dose was present to oxidize S2O32−.
Operation of RSSCTs with concurrent perchlorate/thiosulfate spiking and chlorine oxidation
During RSSCT operations, one pump supplied water that was spiked with the ClO4− and/or S2O32−, while a second pump supplied the HOCl solution separately (when included). This was done to control the contact time between the free chlorine and S2O32− solution. Collapsible plastic containers were used to store the solutions to ensure that the S2O32− did not oxidize before use and that the HOCl did not dissipate before use. Also, the S2O32− solution was replaced every 3 days, so as to maintain its proper concentration. Both solutions were covered with a dark plastic wrap to also ensure no reactions with light.
Various concentrations of chlorine, S2O32−, and ClO4− were appraised while employing deionized distilled water or Redlands, CA, groundwater. Spiked concentration of S2O32− included 40, 250, 500, and 1,000 μg/L as S2O32−. The chlorine dose ranged from 0 to 3.5 mg/L as Cl2 (Table 2), and this was paced to offer either the stochiometric amount needed to oxidize the S2O32− per Equation 1 (in most cases), or to offer an excessive amount.
About 2.2 mg/L chlorine mixed with 0.4% surfactant solution during preloading.
Italicized RSSCTs employed earlier-sampled Redlands water, with 52 μg/L ClO4− and 10 mg/L nitrate as NO3−.
Perchlorate did not breakthrough by 28,000 bed volumes the duration of these tests.
RSSCTs, rapid small scale column tests.
These experiments employed a mixing chamber when reacting the S2O32− with HOCl. The chamber had a 5 min detention time with a stir bar mixing at 250 rpm, which provided a velocity gradient G of 348 s−1. Preliminary mixing tests showed that this achieved sufficient mixing to achieve S2O32− oxidation before the water flow entered the column. To confirm this, the authors tested for the presence of thiosulfate after the 5 min retention time, and found that no S2O32− remained.
Effluent water samples were collected two times per day from the RSSCT effluent until at least 50% of the influent ClO4− concentration was observed in the effluent. The Redlands groundwater hosted a pH of 7.5–8.5, and the RSSCT effluents also were in this range. The deionized distilled water hosted pH values of 6.2–6.8.
Monitoring perchlorate, thiosulfate anion, and surfactants
Perchlorate and S2O32− were monitored by use of Dionex 120 ion chromatograph system, which was equipped with an AS40 auto sampler, a 4 mm AS16 column, and a 4 mm AG16 guard column. The DS4 suppressor was set at 300 mA and a temperature of 35°C. The ion chromatography (IC) protocol employed an eluent sodium hydroxide solution of 25 mM (if not otherwise identified), or 30 mM (when specifically identified herein). When 25 mM of NaOH was employed, the relative peak time for thiosulfate ranged between 7.0 and 7.8 min, while the perchlorate peak times ranged between 14.0 and 15 min. Shorter peak times were observed at a higher eluent concentration: at 30 mM, the thiosulfate peak ranged from 5.5 to 6.1 min, and the perchlorate peaks ranged from 10 to 11.2 min. Peak time also varied with column age. Other anions were also monitored with Dionex 120 ion chromatograph system, while using the eluent concentrations that were best suited for those species. Since thiosulfate and other intermediate sulfur species can be unstable, their analyses were processed through the IC immediately after sampling.
The concentrations of surfactants were monitored by a colorimetric method (Tsubouchi et al., 1981; Parette et al., 2005). This method has a detection limit of 0.1–0.2 mg/L.
Results and Discussion
Initial comparisons of fresh versus aged Redlands water
In early work that compared various RSSCTs and field scale operations, the team had observed variations in bed life performance. Specifically, some of our early work employed freshly received Redlands water for RSSCTs; that is, this water was used within 1 week of its receipt in a 55-gallon sealed barrel. When using this fresh water, the ClO4− broke through considerably sooner than when RSSCTs employed Redlands water that had been aged (stored) for longer than a month (Fig. 1). Moreover, the initial breakthrough for the full-scale Redlands demonstration occurred at about the same time as it did for the RSSCT with fresh Redlands water (Fig. 1). These initial RSSCTs and full-scale demonstrations used the earlier-sampled Redlands water, with ClO4− at 52 μg/L and nitrate at 10 mg/L as NO3−; and the surfactant tailoring agent was tallowalkyltrimethylammonium chloride (Arquad T-50). Some of these differences could be attributed to inherent distinctions between RSSCTs and full-scale operations. For example, others have observed that RSSCTs can often exhibit sharper breakthrough profiles than do full-scale beds, and that was the pattern observed here, where the full-scale bed exhibited earlier initial breakthrough, but the same BV to half breakthrough as did the RSSCT. Also, some sorbing compounds behave according to proportional diffusivity, whereas others do not (Patterson, 2009). Also, the field-scale GAC hosted slightly less surfactant loading—by 10%–15%—than did the RSSCTs.

Comparison of rapid small scale column tests (RSSCTs) with fresh versus aged Redlands water and with full-scale performance. Tests used Redlands groundwater with 52 μg/L ClO4− and 10 mg/L nitrate as NO3−.
Nonetheless, the distinctions between the RSSCTs with freshly collected water versus RSSCT with aged Redlands Water had to be attributed to something other than similitude considerations. The authors hypothesized that these distinctions were associated with competitive oxyanions whose concentrations were redox sensitive. For the conditions herein, we particularly suspected sulfur-based oxyanions that were intermediate between sulfide and sulfates. As a substantiation of this hypothesis, ion chromatograms of fresh Redlands water exhibited peaks that just preceded the ClO4− peak; these peaks disappeared in the aged Redlands Water. For example, when the raw Redlands groundwater was collected into a sealed vial and then immediately mailed overnight to Penn State, our team observed a distinct ion chromatograph peak at 6.1 min (Fig. 2), and this corresponded to the S2O32− peak for the column conditions and 30 mM NaOH eluent that was employed at that time. By conducting ion chromatogram standards via protocols that matched those for this sample (but not on the concurrent day), the authors surmised that this thiosulfate peak in Fig. 2 corresponded to ∼20 to 100 μg/L. The Fig. 2 response curve also showed perchlorate peaking at 11.2 min, which also conformed to the results with a 30 mM NaOH eluent.
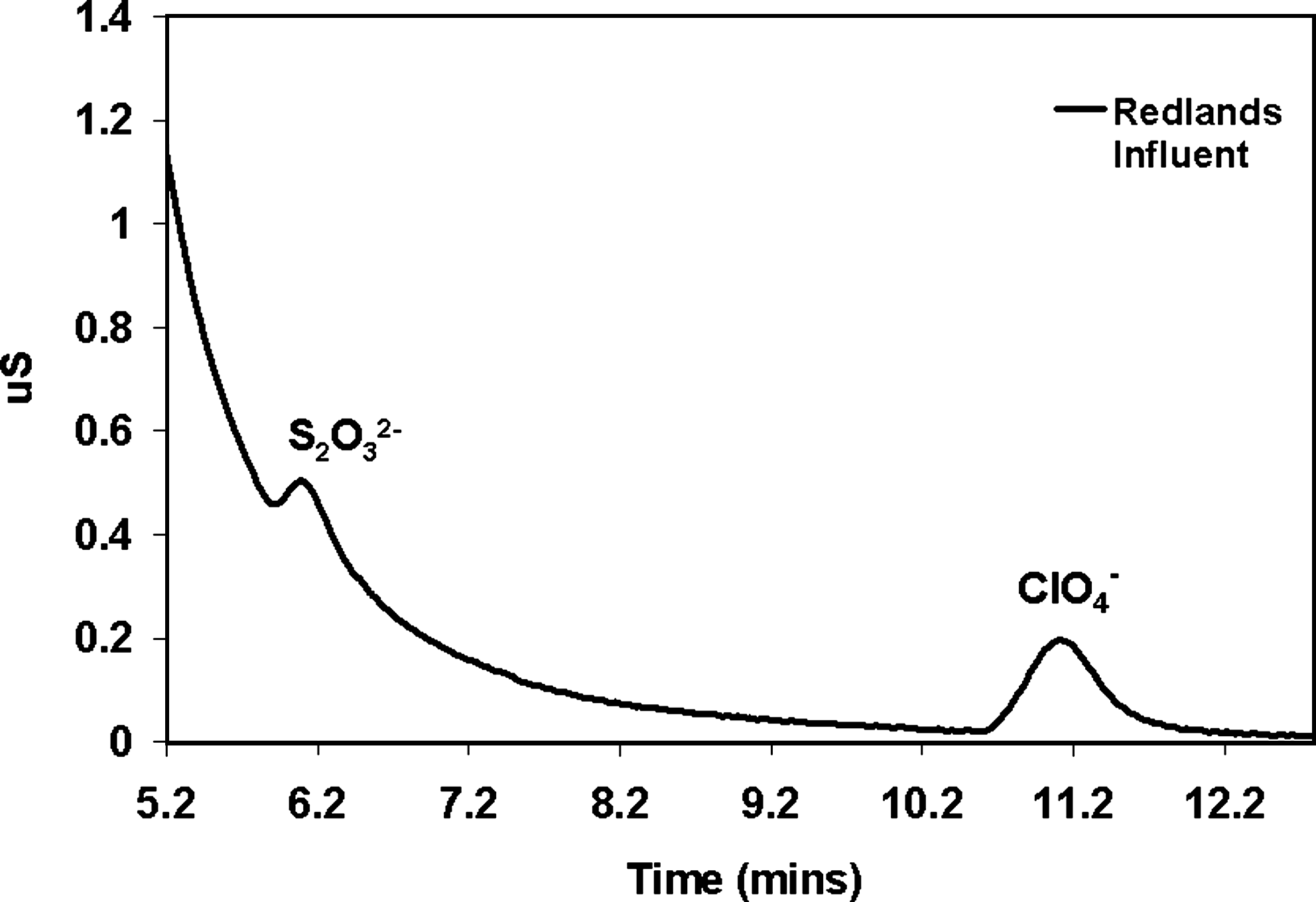
Conductivity chromatogram for Redlands groundwater that was sampled on-site and shipped overnight to be tested. Chromatogram shows retention times of ClO4− and a then-unknown peak that corresponded to when thiosulfate would elute using the DX 120 ion chromatogram instrument with 30 mM NaOH eluent.
From an ion exchange treatment perspective, these peaks occur just before the ClO4− peak. Since ion chromatogram resin media often employ quaternary ammonium functionality, this similar time to peak breakthrough behavior inferred that likewise in the quaternary ammonium-tailored GACs, these same intermediate sulfur species would likewise compete with ClO4− relative to RSSCT bed life.
Stability of sulfur species in the presence of incidental air contact
The authors sought to discern the stability of the reduced sulfur species via two protocols. First, the authors spiked 1 mg/L S2− into deionized/distilled water that was held within a container that was either (1) closed but not air tight, or (2) open to air on top with no lid. These two configurations yielded the same results. Within an hour exposure to this incidental air contact, a fraction of the S2− oxidized to SO42−—to a concentration of 600 μg/L, and the concentration increased slightly to 620 μg/L (210 μg/L as S) after 3 days (Fig. 3). More significantly, the thiosulfate peak also prominently increased in concentration over the 3-day period. Concentration of S2O32− increased from 220 μg/L observed on day 1 to 340 μg/L (200 μg/L as S) on day 3. Thus, roughly half of the sulfide had oxidized to these two species in 3 days. These analyses employed 25 mM NaOH eluent, for which the IC thiosulfate peak generally appeared at 7.0–7.4 min. Additionally, the chromatogram showed two other peaks that exited the IC after the S2O32− had, and these were taken to be other intermediate valence sulfur species (Fig. 3). Next, the authors also sought to discern how long the spiked S2O32− would remain in a stock water supply. For this test, 1 mg/L sodium thiosulfate was spiked into either distilled water or Redlands groundwater. These solutions remained in a closed 5-gallon container that allowed for incidental surface contact with air, but no mixing. After 3 days the S2O32− concentration in solution decreased by a mere 5%, at day 4 by 20%, and after 5 days by 50%. During these tests, the average dissolved oxygen concentration was 1.0. In light of this trial, the thiosulfate solutions used in subsequent RSSCTs were changed every 3 days.
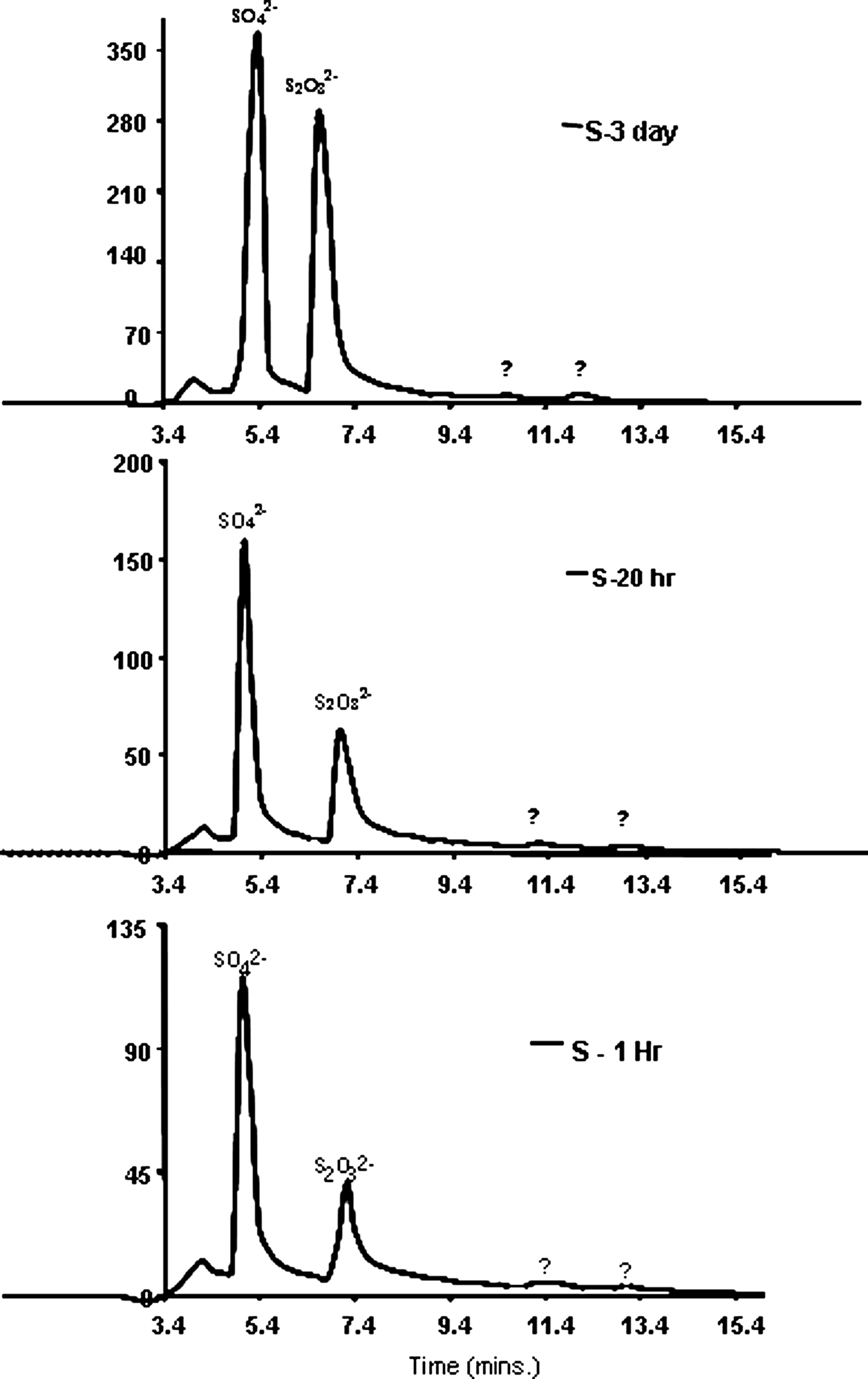
Conductivity chromatogram, exhibiting (relatively) sulfide oxidation via incidental contact with air, after 1 h, 20 h, and 3 days, while using 25 mM NaOH eluent. Initial concentration was 1 mg/L of sulfide in deionized distilled water.
Competition by thiosulfate for perchlorate sorption and chlorine oxidation
The first array of RSSCTs employed deionized distilled water that was spiked with 1,000 μg/L ClO4− plus various levels of S2O32− (0–1,000 μg/L) and free chlorine (0–3.5 mg/L), as presented in Table 2. When 1,000 μg/L ClO4− alone was present in deionized distilled water, surfactant-tailored GAC removed ClO4− to below 6 μg/L for 15,000 BVs. However, when the deionized distilled water also contained 1,000 μg/L S2O32−, the tailored GAC removed ClO4− to below 6 μg/L for only 8,000 BVs (Fig. 4). Thus S2O32− did indeed compete with ClO4− for adsorption sites.
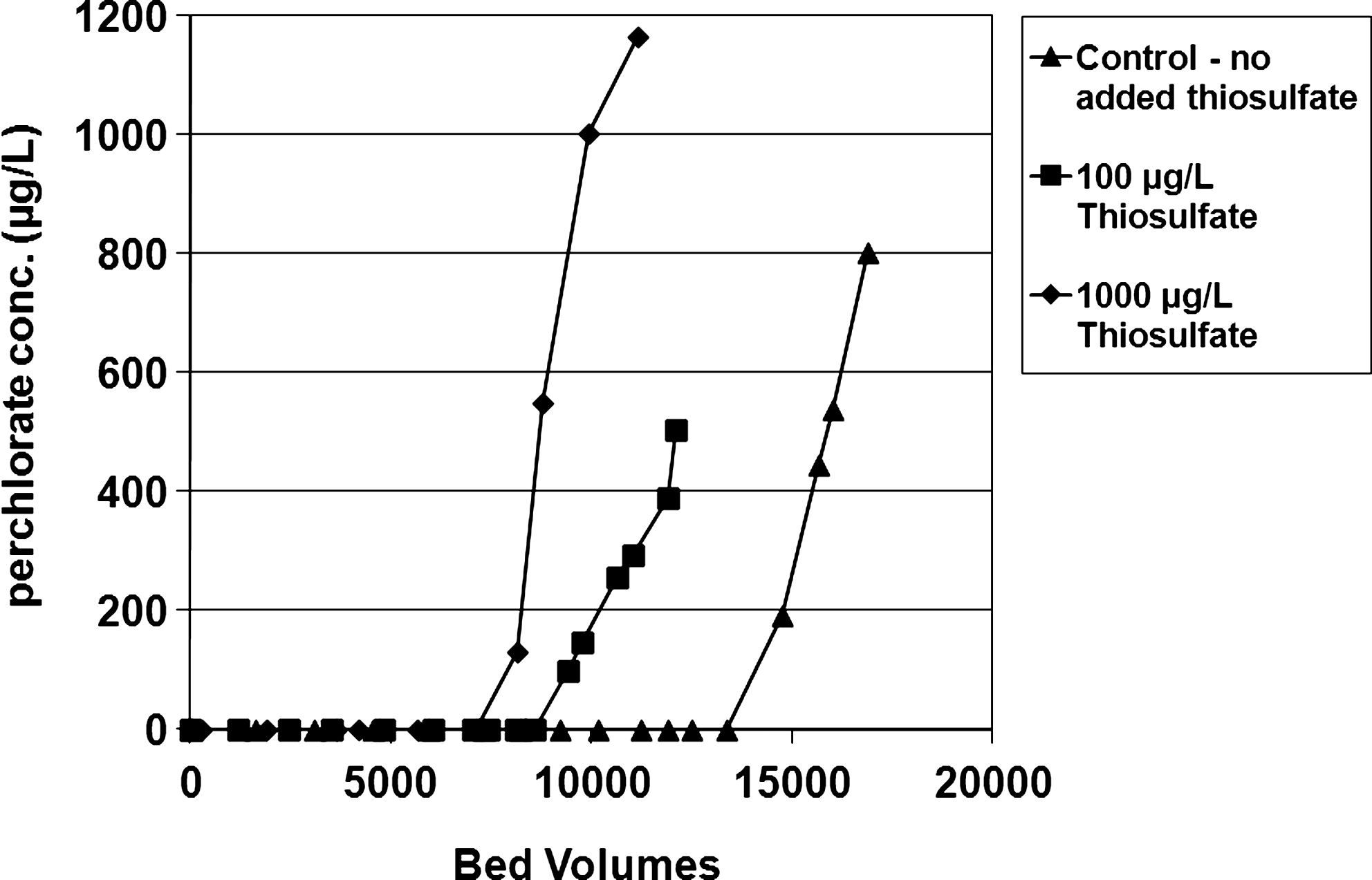
Effects of S2O32− on ClO4− adsorption to surfactant-tailored granular-activated carbon (GAC) using deionized distilled water. All samples included 1,000 μg/L ClO4−. Control is with no S2O32−.
Next, the authors sought to discern the effects of adding 2,500 μg/L (i.e., 2.5 mg/L) chlorine to deionized distilled water that contained 1,000 μg/L ClO4− but no S2O32−. In this case, the chlorine diminished bed life to 9,000 BV. The authors perceived that the chlorine had oxidized the carbon surface, but not the quaternary ammonium functionality (refer to Table 2 line 6) (Patterson, 2009). Then, when the chlorine oxidized the carbon surface it rendered it with a more negative charge, which imposed a reverse Donnan effect (Jang et al., 2009) that precluded the perchlorate from penetrating the pores to where the resident surfactant quaternary ammonium could sorb the perchlorate.
The authors next conducted three experiments with deionized distilled water that included 1,000 μg/L ClO4−. For the control (spiked with only ClO4−), breakthrough occurred at 15,000 BV. However, when 100 μg/L of S2O32− was also included, 6 μg/L breakthrough occurred at 9,500 BV (Fig. 5). In contrast, when the stoichiometric amount of 0.25 mg/L chlorine was used to oxidize the S2O32− to SO42− (per Equation 2), bed life reverted to 13,000 BV. Thus, this prechlorination process sustained the capacity of GAC to remove ClO4− when the spiked S2O32− was oxidized by the chlorine. SO42− competed far less than did S2O32− with ClO4− in this surfactant-tailored GAC as it processed native groundwaters (Patterson, 2009; Patterson et al., 2010). Specifically, as presented in Patterson et al. (2010), we observed that whether a natural groundwater contained 5 mg/L sulfate or was spiked with 50 or 250 mg/L sulfate, the BVs to ClO4− breakthrough did not change much. This result reflected several opposing trends: on the one hand, the higher total dissolved solids (TDS) of the sulfate incurred lower activity coefficients for multivalent species such that they did not compete with ClO4− as extensively. On the other hand, sulfate, with its divalent charge, could exhibit some competition with ClO4−. However, the tightly knit quaternary ammonium functionality within in the surfactant micellar structure apparently posed some steric hindrance to sulfate, with its high hydration energy and waters of hydration.
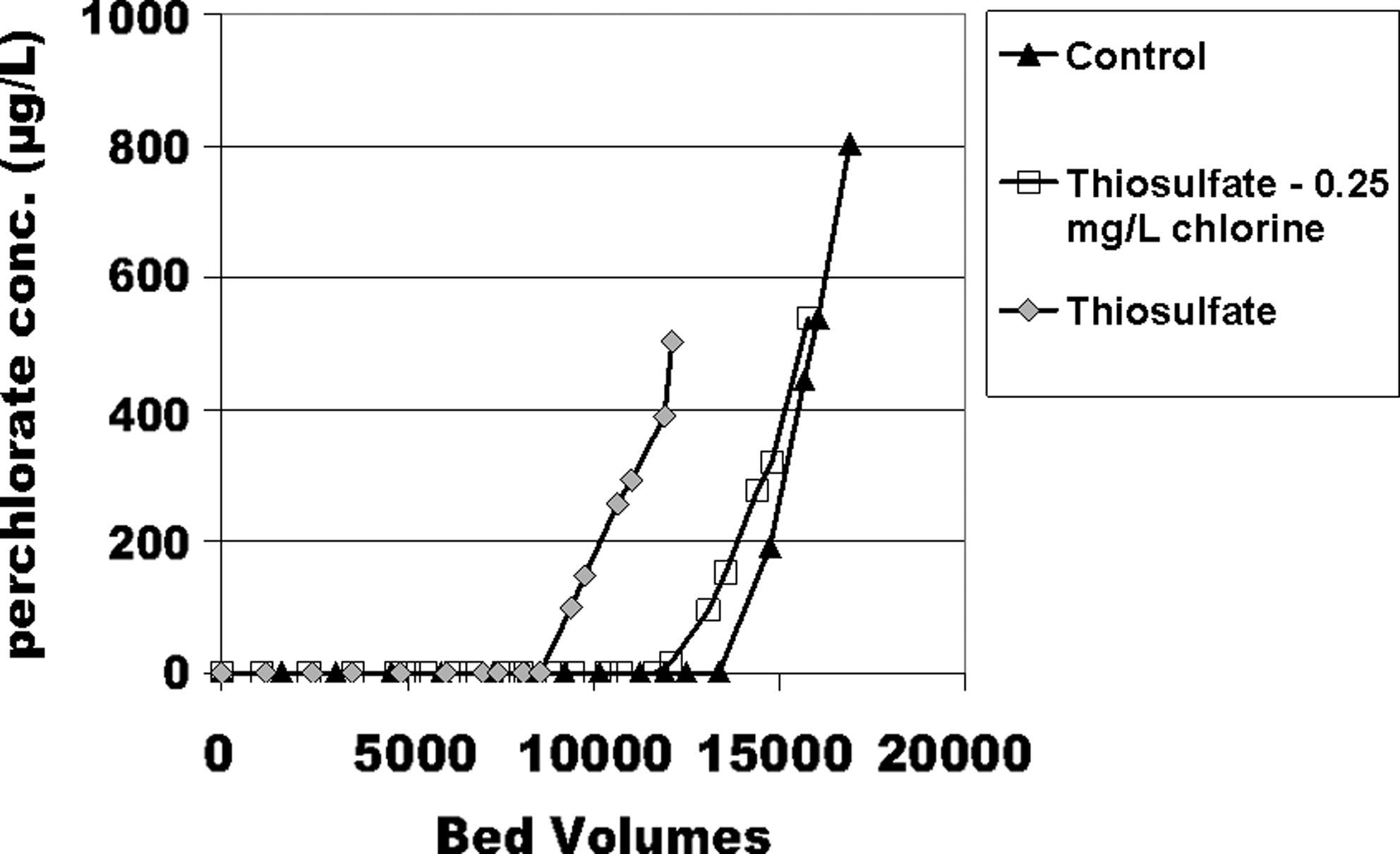
Effects S2O32− concentration and prechlorination on ClO4− adsorption. Influent concentration is 1,000 μg/L ClO4− and 100 μg/L S2O32−, with deionized distilled water.
The significant (and original) finding herein was that the S2O32− competition for ClO4− was considerably greater than the sulfate competition when using these surfactant-preloaded activated carbons.
Redlands groundwater
Next, the authors tested this same chlorination strategy using groundwater from Redlands, CA. The first of these Redlands experiments involved validating the fresh-versus-aged distinctions in RSSCT performance that was discussed above relative to Fig. 1, while using the most recent Redlands water source that contained 38 mg/L nitrate as NO3− and 30 μg/L ClO4−. As shown in Fig. 6, this very fresh Redlands groundwater exhibited 6 μg/L ClO4− breakthrough at 24,000 BV. In contrast, this water after aging (by storage in barrels for more than a month) exhibited breakthrough at 33,000 BV. This result was quite similar to the Fig. 1 trends discussed above (i.e., 22,000 BV and 35,000 BV, respectively). Next, the authors sought to mimic the hypothesized effect of the sulfur intermediate oxyanions by adding 1,000 μg/L S2O32− to aged Redlands water. As observed in Fig. 6, the 6 μg/L ClO4− breakthrough occurred at 25,000 BV (results duplicated). This was virtually the same as when fresh Redlands groundwater had been used. This result concurred with the notion that when the Redlands groundwater was fresh, it contained sulfur oxyanion intermediates species that exerted the same level of competition as 1,000 μg/L of S2O32−. The aged Redlands water was also spiked with 40 and 250 μg/L S2O32−, and even just 250 μg/L S2O32− exerted competition for perchlorate sorption (Fig. 7). In all these RSSCTs, the authors never observed S2O32− presence in the tailored GAC effluent.

Comparing effects of ClO4− adsorption to surfactant tailored GAC when spiked with S2O32− using Redlands water that was aged versus Redlands groundwater that was relatively fresh.
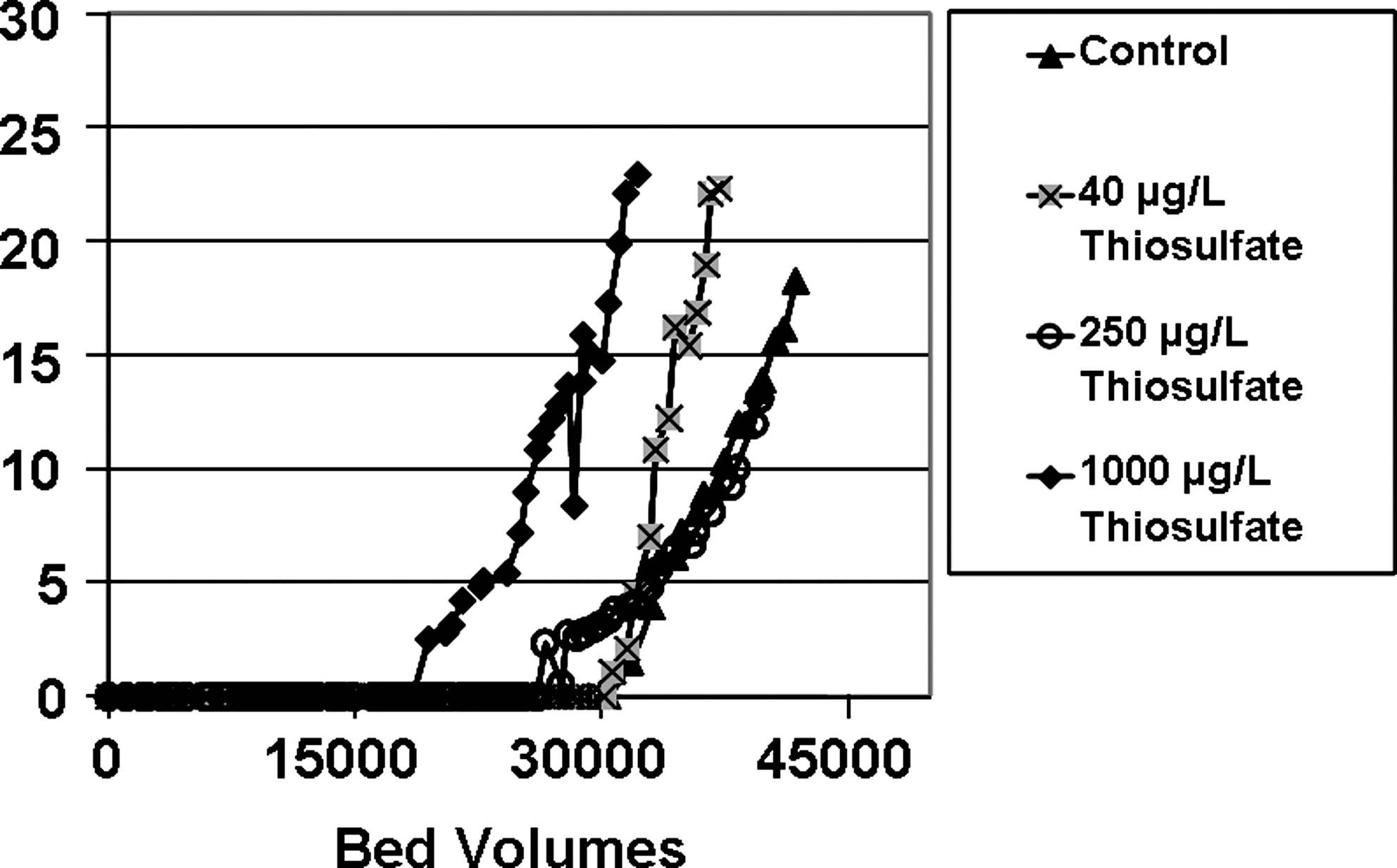
Effects of ClO4− adsorption to tailored GAC when spiked with 40, 250, and 1,000 μg/L of S2O32−, when using Redlands water where the native ClO4− influent is 30 μg/L.
The authors also appraised the effects of chlorination on the oxidation of S2O32− to SO42− were observed (Fig. 8). These RSSCTs were all conducted with aged Redlands groundwater that contained 38 mg/L nitrate as NO3− and 30 μg/L ClO4−. With no prechlorination, the BVs to ClO4− breakthrough was 33,000 BV. The capacity was further diminished by 30% when 1,000 μg/L of S2O32− was spiked into this groundwater. However, when the stochiometric amount of 2.5 mg/L chlorine (needed to oxidize S2O32− per Equation 1) was mixed in, the BV to 6 μg/L ClO4− breakthrough increased to 31,000 BV. Thus, the chlorine again diminished the S2O32− competition by oxidation.

Effects of S2O32− and chlorine on the adsorption of ClO4− to surfactant-tailored GAC when using Redlands groundwater. Native influent ClO4− 30 μg/L. Spiked with 1,000 μg/L S2O32−.
Similar favorable results were observed when 500 μg/L of S2O32− was oxidized by completely mixing with chlorine. Specifically, spiking with a mere 500 μg/L S2O32− diminished the capacity of surfactant-tailored GAC to remove ClO4− down to 17,000 BV. However, prechlorination restored that capacity for the duration that these runs proceeded (Table 2).
In full-scale applications of this technology, the stoichiometrically proper dose of chlorine could be passed to occasional real-time monitoring of S2O32−.
The authors would like to briefly point out that vigorous mixing of chlorine with the water source was important. It was observed that there was no improvement in the surfactant-tailored GAC performance to remove ClO4− when added chlorine was merely feed into the thiosulfate-containing water without such vigorous mixing (Patterson, 2009).
Summary and Conclusions
In RSSCT experiments with GAC that had been preloaded with quaternary ammonium surfactants (TGAC), results showed that thiosulfate greatly diminished the effectiveness of this tailored GAC to remove ClO4− from deionized distilled water or from Redlands groundwater. Also, when thiosulfate concentration equaled 1,000 μg/L, the extent of ClO4− interference was comparable to the diminished ClO4− bed life that has been observed with fresh Redlands groundwater in RSSCTs. Moreover, chlorine oxidation of the thiosulfate increased the bed life of the carbon to adsorb ClO4− when the chlorination was conducted in the proper stochiometric proportion and with complete mixing. However, when the chlorine was added in an unbalanced and incompletely mixed manner such that HOCl or OCl− entered the GAC vessel, this chlorine could react with the carbon surface in an unfavorable manner. Thus, stoichiometrically applied chlorination with proper mixing offers a technically useful means of oxidizing thiosulfate so that this species would not compete as extensively with ClO4− for adsorption to surfactant-tailored GAC. The redox reaction of thiosulfate with chlorine occurred in times measured in minutes and could be achieved via high velocity gradients, which could be achieved at full-scale with in-line blenders or static mixers.
Footnotes
Author Disclosure Statement
No competing financial interests exist.