
Abstract
Abstract
This study systematically compared the evolution of a wide selection of dioxin-relevant compounds during thermal extraction (TE) of filter dust in helium and during de novo (DN) tests in a flow of synthetic air. TE yielded a wide range of dioxin-relevant compounds; however, chlorinated aromatics appeared only in minor amounts. Laboratory DN tests were conducted on this same dust, and also after a mild, medium, or severe TE-procedure, involving final TE-temperatures of 300°C, 420°C, and 540°C, respectively. The same compounds were again analyzed. Chlorinated compounds (Cl-compounds) became predominant DN-products. For each DN-test and for each product, it was verified as to how far preliminary TE and consecutive DN each contribute to the sum of (TE + DN) products. Therefore, for each test, an individual TE- and DN- contribution level could be assigned to each compound monitored. Most oxygenated-compounds were TE-derived, suggesting that they formed from pre-existing oxygenated carbon structures, rather than from oxidation of carbonaceous matter. Conversely, Cl-compounds were typical DN-products and selectively gained importance as TE became more severe. The tests generated much more chlorobenzenes than chlorophenols, in line with the high polychlorinated dibenzofuran to polychlorinated dibenzo-p-dioxin ratio found by conventional analysis of the dust tested. The tests were designed to compare DN- and TE-products and, thus, distinguish between dioxin formation by the precursor route (i.e., formation from structurally related compounds) and the DN route (i.e., low- temperature, catalytic oxidation of the carbonaceous matrix). There was substantial difference between TE- and DN-products, the former being lean in Cl-compounds and the latter being rich. The sample tested was particularly rich in volatile matter and, hence, atypical. The procedure followed stepwise eliminates this volatile matter, so that DN-products from the original sample could be compared with those in which fixed carbon is more important. Preliminary TE strongly enhanced the relative importance of Cl-compounds and reduced that of oxygenated and hydrocarbon compounds during the consecutive DN tests.
Introduction
The oxidation of carbon and its link with dioxin formation has been demonstrated in a recent work (Grandesso et al., 2008). The implications for incineration and other thermal processes have been reviewed (McKay, 2002; Stanmore, 2004; Alderman, 2005). Precursor theory explains the formation of dioxins in thermal processes on the basis of structural similarity with precursors, for example, polychlorinated phenols (PCPh), biphenyls (PCB), benzenes (PCBz), and of experimental evidence of their conversion into polychlorinated dibenzo-p-dioxins and dibenzofurans (in short, dioxins or PCDD/F). Also, polycyclic aromatic hydrocarbons (PAH) can be converted into dioxins albeit that—depending on their structure—PCDD/F-yields span at least four orders of magnitude, from 160 ng/g phenanthrene to 1.42 mg/g biphenyl (Wilhem et al., 2001; Fullana and Sidhu, 2005). Significant amounts of PCDD can be generated from a cocktail of organic compounds (Cieplik et al., 2003), yet with insufficient associated PCDF to create the typical thermal fingerprint. There is evidence that PCDF mainly derive from either PAH or carbonaceous matter, single aromatic nuclei failing to scramble (Hell et al., 2001), whereas PCDD originate from both chlorophenol condensation and DN formation. This shows the merits of a wider analytical effort, identifying not only PCDD/F but also potential precursors and surrogates. The latter may be sampled from flue gas or—as in Tsytsik et al. (2010)—identified among the products desorbed during lab-scale testing.
Dioxin formation is based on incomplete low-temperature catalytic combustion of carbon, yielding also a wide range of other products of incomplete combustion. This pathway has been repeatedly demonstrated using both synthetic matrices and plant-derived dust. Some researchers thermally or solvent cleaned the original fly ash with the purpose of demonstrating dioxin formation from precursor-free fly ash, but their experiments remained inconclusive (Albrecht and Karasek, 1995). However, dioxin laboratory tests fail to present the level of activity required to explain the levels of dioxins observed (Nordsieck et al., 2001). This underperformance could be linked to the hyperactivity of freshly formed samples that is lost when aged. Another explanation is that long residence times in deposited dust compensate this deficit in activity, explaining at the same time the “memory effects” in slowly converting deposits.
During DN tests, a wide range of products is formed, starting from suitable structures in fly ash (Schwarz and Stieglitz, 1992). Jay and Stieglitz (1995) identified 250 organic compounds in flue gases from municipal solid waste incineration, mainly hydrocarbons, halogen-compounds, esters, aldehydes, ketones, carboxylic and hydroxyl-compounds (including chlorophenols). The ratio of aliphatics (not covered in this work) to aromatics was 40/60, except for halogen-compounds, where aromatics only represent 14%. Only few authors monitored numerous other compound classes (Takasuga et al., 1994); the relative importance and distribution of specific compounds seem to depend on the system studied. For Schwarz and Stieglitz (1992), chlorobenzenes represent the bulk of the output; for Takasuga et al. (1994), chlorophenols are more prevalent.
In our research, emphasis is on industrial scale testing, designed to test practical measures and solve environmental problems in thermal and metallurgical plants. Industrial-scale testing is severely restricted, however, with regard to the range of feasible testing conditions. To accelerate testing, widen its scope, and reduce expenditure, much time and effort was devoted to the development of appropriate test methods and to the testing of a wide variety of dust samples in the laboratory (Tsytsik et al., 2008).
The DN route requires oxygen, chlorides, and contact materials comprehending catalytic transition metals. In this particular case, an aluminum melting furnace, only oxygen is fully available in the off-gas and carbon, chloride, dust, and catalysts all arise from unintentional impurities adhering to scrap. No flux or chemical degassing agent is used. The off-gas is low in dust, even before the filter. The dust in these tests is rich in soot, PAH, PCDF, and PCB, and the ratio of PCDF to PCDD is high (Tsytsik et al., 2010), suggesting that DN route of dioxin formation prevails in this system, melting clean aluminum scrap.
This article describes some DN test results with a purpose: comparison of DN yields and products from four samples that are distinct in their pretreatment—either raw or after preliminary TE at three levels of severity.
Experimental Section
Materials
The sample tested is a baghouse filter dust, derived from a reverberatory furnace of a clean aluminum scrap melting plant. It is a cohesive black powder, resulting from (unknown) impurities accompanying this scrap. Such samples are poorly reproducible yet interesting for studying the formation of dioxins during the melting of scrap.
Analytical reagents were obtained from Supelco: EPA chlorinated hydrocarbon mix 8120 and 2000 μg/mL in methanol; EPA chlorinated hydrocarbons mix 8121 and 2000 μg/mL, in methanol; EPA 525 PAH mix, 500 μg/mL in dichloromethane; 200 μg/mL in methanol. A benzene, toluene, ethylbenzene, xylenes (BTEX) mixture, 200 μg/mL in methanol was purchased from Restek; decafluorobiphenyl solution (10 g/L) was prepared from decafluorobiphenyl powder (Aldrich Chemical and diluted in methanol. Methanol (purge and trap grade) and dichloromethane (gas chromatography [GC] grade, SupraSolv) were from Merck.
Preliminary sample analysis
Before this testing program involving both TE and DN tests, numerous analyses and tests were conducted with the purpose of first selecting and then characterizing this particular sample. The oxidation of carbon in the sample was continuously monitored in airflow, examining its heat effects (differential thermal analysis [DTA]) and its weight loss (thermogravimetric analysis [TGA]) combined with on-line analysis of carbon dioxide and monoxide, water vapor (mass spectrometry [MS] in full scan mode), and organics (MS in multiple ion monitoring mode) from the sample (Fig. 1). These results will be discussed in a dedicated paper.
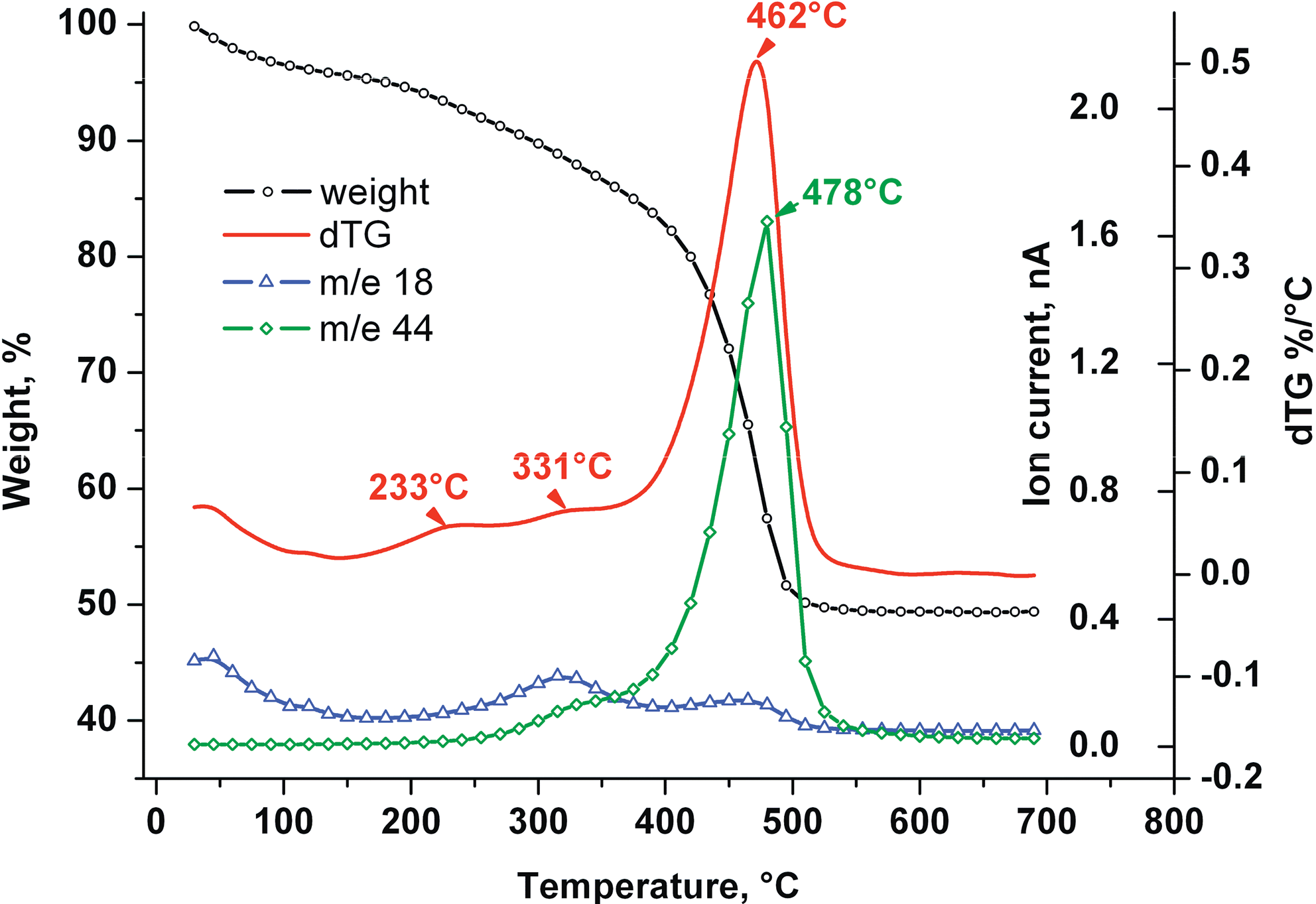
Thermogravimetric analysis-graph showing the evolution with temperature (and time) of sample weight, its time derivative differential weight evolution in ThermoGravity (dTG), and the mass spectrometry signals for m/e 18 (water vapor) and 44 (carbon dioxide). m/e, mass-to-charge ratio.
TE and DN-test
All DN-tests were performed on either a fresh or a previously thermally extracted-sample in a thermobalance (951 Thermogravimetric Analyzer; TGA, DuPont Instruments), trapping evolving products on a sampling tube filled with sorbent. The latter was analyzed by thermal desorption (TD), followed by GC-MS (TD-GC-MS), resulting in an estimate of the compounds released in the gaseous phase. The same method applies to both TE- and DN-samples, as described elsewhere (Tsytsik et al., 2008).
The TGA runs operated with selected and flexible temperature-time programs adapted to the desired treatment, incorporating both preliminary TE-tests and successive DN-tests. Three levels of TE were tested, corresponding to thermal treatments with endpoints at 300°C, 420°C, and 540°C. DN tests were performed with a standard: gradual heating at 4°C/min to 420°C (DN3 to 450°C). Helium (50 mL/min) was used as carrier gas for TE, and dry air (50 mL/min) was used for DN-tests. This creates the principal difference between TE and DN- tests, that is, neutral and oxidative conditions. TE is described in detail in (Tsytsik et al., 2010), whereas experimental details about the DN tests are given in (Tsytsik et al., 2008).
The entire testing program totals four DN-test sequences combined with 10 preliminary TE-tests, the latter discussed in (Tsytsik et al., 2010). The experiments were designed so that both TE and DN could be evaluated:
DN1, room temperature–420°C, 4°C/min. No preliminary TE1. Reference sample. Mild: TE2 [300°C × 8 min; 300°C × 16 min; 300°C × 32 min]→ DN2, 250°C–420°C, 4°C/min. Medium severity: TE3 [320°C × 48 min; 420°C × 30 min]→ DN3 220°C–450°C, 4°C/min. Severe: TE4 [320°C × 16 min; 360°C × 16 min; 420°C × 16 min;480°C × 32 min; 540°C × 16 min] → DN4, 250°C–420°C, 4°C/min.
Sampling the TGA effluent and TD-GC/MS analysis
Two sequential beds of sorbents Tenax TA and polydimethylsiloxane, inserted inside a glass sampling tube, were used for sampling compounds of vastly different volatility (benzene to benzo[a]anthracene) from the effluent. While sampling, the tubes were kept at −22°C in a mixture of NaCl/ice (1:3).
The sorbate was analyzed in a dedicated TD system (Markes Unity TD) connected to a Finnigan Trace GC Ultra/Trace DSQ mass-spectrometer. Each sampling tube was thermally desorbed at 300°C for 30 min in a continuous flow of helium carrier gas (70 mL/min); the effluent was cryo-focused at −8°C on a graphitized carbon cold trap before subsequent GC/MS analysis. Instrumental settings, operating conditions, and validation of the TD-GC/MS have been described elsewhere (Tsytsik et al., 2008). Analytes were identified by their mass spectra using National Institute of Standards and Technology Mass Spectral Library, 1998. Chromatographic retention times were used in some cases to confirm identification.
Quantification was performed by the external standard method. One-point calibration was applied by spiking standard solutions of PCBz, PAH, and BTEX onto separate adsorption tubes, which were thermally extracted in sequence with the tubes used for sampling the effluent. Quantification was based on peak areas of the most abundant ion in the spectra for each target compound. The analytes not present in the calibration solution were semi-quantified by using response of 1,3,5-trichlorobenzene as a reference: 1,3,5-trichlorobenzene was selected as showing the best correlation with the responses of other tested analytes during preliminary technique-validation studies. Applying the same quantification reference for “nonstandard” analytes in all tests allowed generating comparative data.
Recovery was estimated by spiking each sample tube with internal standard (decafluorobiphenyl) before introduction into the TD-GC/MS unit. Carry-over inside the instrumental transfer lines was routinely checked by running instrumental blanks (empty glass tubes) in between the sample runs.
TGA rinses
Not all semi-volatile compounds reach the sampling tube. Analyte losses inside the thermobalance housing and duct to sorbent tube were recovered by rinsing with dichloromethane and subsequent GC-MS analysis as described in (Tsytsik et al., 2008).
Target compounds
The following analytes were selected on the basis of their structural similarity to dioxins as well as citation in other studies, for example (Schwarz and Stieglitz, 1992; Takasuga et al., 1994):
(a) BTEX, or TEX if benzene was not analyzed), biphenyl, and naphthalene. These are the most volatile compounds monitored. (b) Cl-compounds, namely (i) mono- to hexachlorinated benzenes (chlorobenzenes, PCBz), (ii) mono- to pentachlorinated naphthalenes (polychlorinated naphthalenes [PCN]), (iii) mono- to trichlorinated biphenyls (PCB), (iv) mono- to pentachlorinated benzonitriles (polychlorinated benzonitriles [PCBzN]), (v) di- and tetra- chlorinated thiophenes (polychlorinated thiophenes [PCTh]), and (vi) mono- to pentachlorinated phenols (chlorophenols, PCPh). (c) PAH, limited to acenaphthylene, acenaphthene, anthracene, benzo[a]anthracene, benzo[a]pyrene, benzo[k]- and [b]-fluoranthene, chrysene, fluorene, phenanthrene, and pyrene. (d) Oxygenated compounds (O-compounds), that is, acetophenone, benzaldehyde, benzofuran, benzophenone, benzoic acid, dibenzofuran, fluorenone, phenol, and xanthone. (e) Sulfur-containing compounds (S-compounds) thiophene, benzothiophene, and benzothiophenone. (f) Nitrogen-compounds (N-compounds) BzN, benzenedicarbonitrile, acridine/benzoisoquinoline, isoquinoline/quinoline, and naphthylamine were tentatively identified.
In further discussions, biphenyl, naphthalene, BTEX, and PAH are briefly referred to as “aromatics,” excluding the aromatic Cl-, O-, S-, and N-compounds defined earlier.
Results and Discussion
Preliminary sample analysis
The oxidation of “carbon” is representative for DN-activity and was monitored during separate tests. DTA (while heating a dust sample at 10°C/min to 800°C in air, 150 mL/min) shows successive exothermal carbon oxidation domains, peaking at 330°C and 472°C, respectively. The first corresponds to the low-temperature, catalytic conversion, characteristic for DN reactions; the second and most important peak corresponds to the complete conversion in carbon dioxide and monoxide. Parallel TGA-MS (TGA at 10°C/min to 750°C in air, 25 mL/min, combined with mass spectrometric monitoring of evolving volatiles) in air identified three successive ranges of weight loss (Fig. 1); these culminate at 233°C, 331°C, and 462°C (comparable to the DTA results) and leave a residual weight of 91, 86, and 50 wt% at temperatures of 273°C, 360°C, and 537°C, respectively. The first domain coincides with striking evolution of PAH and O-compounds (1) and ends at 273°C, the next two domains correspond with potential domains of DN activity. Oxidative breakdown stimulates extensive evolution of both aliphatic and aromatic compounds in the first domain (273°C–360°C). In the second, oxidation is fairly complete.
On-line MS
MS allows on-line monitoring of evolving DN-products. Carbon dioxide evolves already below 200°C (after desorption or decomposition), and its concentration peaks at 478°C. Its evolution can be directly confronted with TGA- (Fig. 1) and DTA-curves. Products of incomplete combustion, that is, carbon monoxide, aliphatic and aromatic hydrocarbons, as well as some oxygenated combustion products and even PCBz were monitored on line. Carbon monoxide evolves in a range of 170°C–570°C, with a marked first peak at 327°C and a small peak at 462°C, in line with aforementioned domains of slow and fast oxidation. Water evolves during drying and combustion. Paraffins and olefins roughly arise from 200°C to 400°C, with a single Gaussian peak at ca. 320°C. Other organics monitored on line are tentatively identified as toluene/xylenes (mass-to-charge ratio [m/e] 91, 92, 106), biphenyl/acenaphthene (m/e 153, 154), phenol (m/e 65, 94), benzofuran (m/e 90, 118), and acetophenone/benzophenone/benzaldehyde (m/e 77, 105, 106). Mono- to pentaCBz were successfully monitored on line. Tentative identifications were confirmed by both the analytical procedures and extraction-based analysis.
The severity of TE, used for “cleaning” the sample before the DN-test, can be described by the TE-temperature reached, that is, 300°C (mild), 420°C (medium), and 540°C (severe). Table 1 shows the total yield (μg/g) of both the TE- and DN-product classes (aromatics, O-, Cl-, and N- compounds). The data (Figs. 2 and 3) suggest complex relationships between TE- and DN-yields and TE-severity. Mild TE slightly inflates the (TE + DN)-output of aromatics, leaving O- and Cl-compounds at standard test (DN1) levels. Medium-severity TE volatilizes huge amounts of O-compounds, much more even than during severe TE, which is remarkable. This medium-severity TE obviously activates the sample, so that the resulting TE and DN values far exceed expectations. The yield of aromatics monotonously increases with TE-severity and forms its best descriptor.
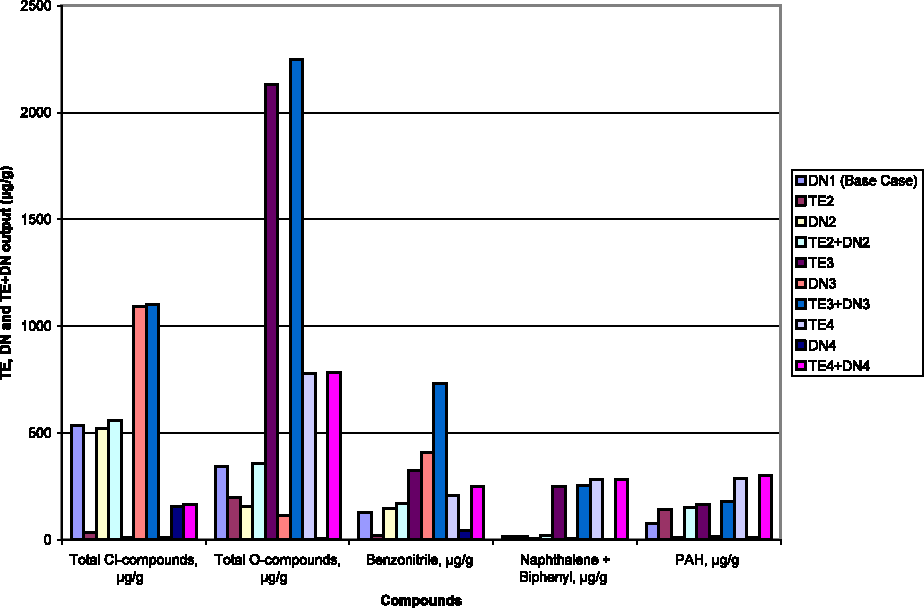
Amount (μg/g) of the different product groups in DN and preliminary TE-tests, followed by their sum TE + DN. DN, de novo; TE, thermal extraction; PAH, polycyclic aromatic hydrocarbons; Cl-compounds, chlorinated compounds; O-compounds, oxygenated-compounds.
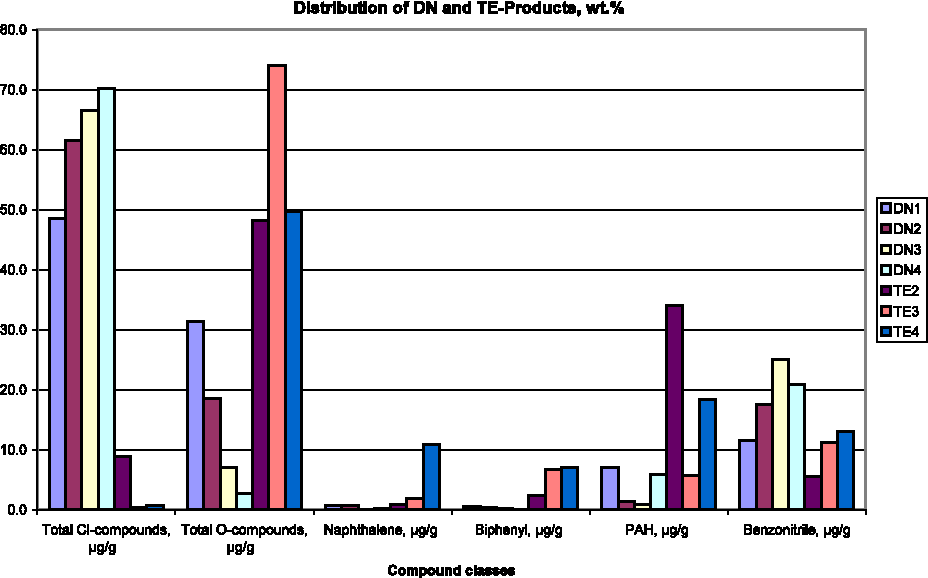
Distribution (wt%) of the different product groups in the DN-tests and the preliminary TE-tests.
Units: μg/g (wt%, relative to the standard test DN1). The highest DN, TE, and (TE + DN)-values encountered are marked in bold, italic bold, and underlined bold, respectively.
Major nitrogen-compound. Other nitrogen-compounds have not been monitored as systematically.
DN, de novo; TE, thermal extraction; O-compounds, oxygenated-compounds; S-compounds, sulfur-containing compounds; Cl-compounds, chlorinated compounds; <DL, below detection limits.
DN-sample activity
DN-sample activity can be described by the yield of evolving Cl-compounds (0.15–1.1 mg/g), to which TE scarcely contributes. Figure 2 compares the output (μg/g) of four DN-tests, that is, raw sample (DN1), or after mild (DN2), medium (DN3), and severe TE (DN4), the cumulative result of three preliminary sets of TE-runs (TE2 to TE4), and the (TE + DN)-values resulting from adding the corresponding TE and DN contributions. DN-activity is similar for raw and mildly extracted samples; it is roughly doubled, however, for medium TE, and after severe TE at 540°C still represents some 30% of original DN1-activity. Volatile matter is then already largely eliminated from the sample, so that presumably Residual carbon becomes the principal source of DN-products. For this particular filter dust sample, the evolving volatile matter strongly marks the output of O-compounds. Among both TE- and DN-products, the rising importance of BzN is remarkable.
Figure 3 compares the output distribution of four DN-tests and the three preliminary TE-procedures. With rising TE-severity, Cl-compounds increasingly prevail, suggesting that carbonized substances preferentially generate Cl-compounds under (oxidizing) DN-conditions. In parallel, the output of O-compounds systematically declines, yet these obviously prevail among TE-products. O-compounds, thus, seem associated with pre-existing, O-containing carbon surface structures, rather than to result from catalytic carbon oxidation, essential in DN formation. This is in line with earlier findings regarding the origins of PCDD/F-oxygen, as established by DN-tests with O18 artificial air (Addink and Olie, 1993; Olie et al., 1995).
Tables 2 and 3 in more detail show the data obtained for the various chlorinated families and for the most important O-compounds.
Units: Amount, μg/g (distribution, wt% of total Cl-compounds).
PCBz, polychlorinated benzenes; PCPh, polychlorinated phenols; PCN, polychlorinated naphthalenes; PCB, polychlorinated biphenyls; PCBzF, polychlorobenzofurans; PCDF, polychlorinated dibenzofurans; PCTh, polychlorinated thiophenes; PCBzPh, polychlorobenzophenones; PCBzN, polychlorinated benzonitriles.
Units: Amount, μg/g (distribution, wt% of total O-compounds).
For each test, the TE or DN-origin of specific compounds can now be verified, by computing a ratio DN/(TE + DN), yielding 100% for the base case (DN only) and a variable value (0%–100%) after mild, medium, or severe TE. The results are reported in Table 4. Cl-compounds are typical DN-products, with as sole exception chlorophenols, representing 76, 21, and 0 wt% of (TE + DN), after mild, medium, and severe-TE, respectively. Conversely, aromatics and O-compounds are almost purely TE-products; only xanthone and fluorenone pay a tangible contribution, of 9, and 7.1 wt%, to the sum (TE + DN).
0 DN value was <DL, whereas the compound was still detected during TE.
<DL Both TE and DN values remained below the limit of detection.
If benzene was not analyzed, BTEX value is given as the sum of toluene, ethylbenzene, and xylenes.
n.a., Not analyzed; BTEX, benzene, toluene, ethylbenzene, xylenes mixture; PAH, polycyclic aromatic hydrocarbons.
The DN-products can, thus, be visualized as products of chlorination, diluted to some extent by evolving volatile matter. For the specific sample tested, the latter are particularly rich in O-products. The latter, however, are readily eliminated by TE-treatment and fail being replaced by oxidative breakdown.
Discussion of the various product classes
Cl-compounds
Nine Cl-compound classes are identified during DN-tests, against only three (PCBz, PCPh, and PCBzN) during TE-tests (Table 2). PCDF remains low key, with a single appearance at ca. 300 ng/g of di-CDF. Both semi-volatility and too high detection limits still limit this type of analysis: In further work, the analytical method applied should be completed with a full range analysis of PCDD/F (P = 1 to 8), PCB, and PCN (P = 1 to 10). The TE-contribution to (TE + DN) varies from nil to 8%, except for chlorophenols (Table 4); PCN, PCB, polychlorobenzofuran (PCBzF), PCDF, PCTh, and PCBzN remained undetected during TE, hence they are to be qualified 100% DN.
PCBz are for 93%–98% of DN-origin. The weight average chlorination level is higher in DN: 2.8 ± 0.1, against only for direct TE and 2.2 ± 0.4 for indirect TE.
PCPh rapidly decline with rising TE-severity. The ratio of PCPh/PCBz both in base case and mild TE is a meager 1.7%, yet it further drops to 0.1% after a medium-severity TE and below detection limits after severe TE. The ratio of mother substances (Ph/Bz) attains 2.5 in the base case and 0.55 after medium-severity TE.
The most typical aspect of DN-activity is its chlorinating capacity, leading to a prevalence of Cl-compounds. Conversely, TE leads to dechlorination, reducing their relative importance. Hence, the ratio of Cl-compounds to their parent molecule is now verified in DN-tests. The ratio of PCBz to (PCBz + benzene) varies between 92% (base case) and 97% (medium TE). The ratio of PCN to (PCN + naphthalene) is less, that is, 58% (base case) and 68%, 93%, and 61% after mild, medium, and severe TE, respectively; PCB to (PCB +biphenyl) leads to low values of 15, 30, 31, and 11 wt%. For PCBzF, these are respectively 13, 31, 43, and 53 wt%. Finally, the values for PCPh/(PCPh + phenol) vary erratically, with 7% (base case), 22% (mild TE), 5% (medium TE), and nil (severe TE).
The total amount of DN-Cl-compounds corresponds to 0.15–1.09 mg/g in the four DN-tests (Table 1), to be compared with 1% of carbon converted to chlorobenzenes and 0.01%–0.04% transformed into dioxins (Cieplik et al., 2003). This suggests that this system is one order of magnitude lower in chlorinating capacity.
O-compounds
Surprisingly, oxidizing DN-conditions fail to stimulate formation of O-compounds, which mainly appear as TE-products (Fig. 1). Phenol is the main O-compound. It is sensitive to preliminary TE, which decreases DN-phenol yield from 29% of O-compounds in the base case to nil, after a severe TE. Together, benzaldehyde and benzoic acid are main DN O-compounds, yet they entirely disappear after severe-TE. Also, acetophenone shows a declining trend for DN-tests with higher TE-severity. Benzophenone, fluorenone, and xanthone are typical DN-products; their selectivity rises with increasing TE. Together with benzoic acid, they are well represented in rinses (Table 3). Benzofuran attains high values in DN-tests after mild and medium TE. Dibenzofuran strongly rises in DN after medium and severe TE and also in TE at 420°C or higher. See Table 4.
N- and S- compounds
BzN is mentioned in surprising numbers of combustion studies. Both TE and DN contribute to BzN (Table 1). Other compounds identified are benzene-dicarbonitrile, acridine/benzoisoquinoline, isoquinoline/quinoline (at levels of 10–60 μg/g), and even a trace of naphtylamine (7 μg/g). Remarkably, chlorinated S-compounds (di- and tetra- CTh) appear, the corresponding mother molecule remaining undetected.
PAH
PAH are chiefly TE-products, with only a 4.3%–8.7% DN-contribution to the sum (TE + DN). The total amount of PAH evolving during TE continuously rises with severity (Table 1). In the base case and after mild TE, the less volatile PAH (with more than three aromatic rings) constitute more than half of DN-PAH. After medium or severe TE, phenanthrene dominates.
BTEX–naphthalene–biphenyl
BTEX, naphthalene, and biphenyl all are basically TE-products. Benzene is the lightest product detected and only quantified in part of the tests. Naphthalene and biphenyl decrease in importance at rising TE-severity (Table 4).
Rinses
During both the preliminary TE-runs and the successive DN-run, semi-volatiles, such as PCDD/F, may accumulate on the TGA-housing and the ducts to the sampling tube. Such laggards are collected after the test and examined. TGA-rinses were examined for the TE2 + DN2 test (Table 5). Therefore, mainly PAH (starting from phenanthrene) and O-compounds are found, implying that their TE- and DN-values are systematically underestimated. Although higher-Cl-compounds were searched for, these remained below detection limits. The most important dioxins surrogate, PCBz, is rather well recovered, with losses affecting only pentachlorobenzene and hexachlorobenzene: the weight average chlorination level of PCBz is 5.27 in the rinses, to be compared with 2.26 (TE) and 2.77 (DN). Less volatile PAH and phenones lose up to 46 wt% of their response in this fraction (cf. Table 5).
Both TE and DN values remained below the limit of detection.
If benzene was not analyzed, BTEX value is given as the sum of toluene, ethylbenzene, and xylenes.
n.a., not analyzed.
Statistical analysis
The four DN tests show a reasonably high level of internal correlation. The best-correlated pair are the tests at medium and high severity (r2 = 0.996), the weakest test pair is the base case DN1 + DN4, after severe TE (r2 = 0.913). The Cl-compounds, believed to typify DN activity, correlate well with BzN (r2 = 0.979) and total DN output. They are uncorrelated, however, with any other group, that is, O-compounds, aromatics, and S-compounds, suggesting that their respective mechanisms of formation are unrelated. The sole other relatively strong correlation among DN-results links aromatics and O-compounds (r2 = 0.876).
As a next step, the cross-correlation within the five compound classes is tested among ten TE and four DN tests, all in one combination, with as results:
Total TE and DN output-values are poorly and relatively evenly correlated with individual compound classes (worst with aromatics: r2 = 0.52, best with BzN: r2 = 0.70). Cl-compounds are well correlated with N-compounds (r2 = 0.942), in practice with BzN, but not with any other compound class. Aromatics correlate well with O-compounds (r2 = 0.848). Despite their only casual appearance, S-compounds still correlate well with O-compounds (r2 = 0.928).
As a general conclusion, statistical analysis shows that the DN-formation of Cl-compounds is independent of other phenomena, those relative to BzN excepted. When only the ten TE-runs are considered, some small shifts occur: Cl-compounds correlate now with O-compounds, S-compounds, and aromatics but no longer with benzonitrile, suggesting that Cl-compounds (and benzonitrile) forming mechanisms are not necessarily identical in TE and DN.
Footnotes
Acknowledgments
The authors thank Prof. Pierre Jacobs (Katholieke Universiteit Leuven, Leuven, Belgium) and Dr. Kees Olie (University of Amsterdam, Amsterdam, The Netherlands) for useful discussions.
Author Disclosure Statement
No competing financial interests exist.
†
This article is dedicated to the late Dr. Ludwig Stieglitz, founding father of the de novo theory.