
Abstract
Abstract
Removal of perchlorate (ClO4−) while using granular-activated carbon (GAC) that had been preloaded with cetylpyridinium chloride was appraised using Rapid Small-Scale Column tests (RSSCT) and pilot-scale demonstration. ClO4− competed with nitrate, sulfate, chloride, and other anions for ion exchange sites on the preloaded carbon. Tests used native groundwater from Fontana, CA, which contained nitrate of 32 mg/L as NO3−, 7 μg/L as ClO4−, and 1.9 μg/L (as U) of uranyl carbonate species (predominantly UO2(CO3)34−) species. RSSCT with Fontana groundwater exhibited ClO4− breakthrough at 25,000 bed volumes. When the same groundwater was spiked to three times the native NO3− concentration, ClO4− breakthrough occurred at 12,000 bed volumes. When the total dissolved solids (TDS) increased from the native 250 mg/L up to 660–810 mg/L TDS, by adding magnesium sulfate, sodium bicarbonate, or sodium chloride, the bed-life for removing ClO4− increased by 10%–30%. This result was attributed to perchlorate's competition with native UO2(CO3)34−, which limited the bed-life for ClO4− breakthrough. Specifically, at higher TDS the computed activity coefficient of UO2(CO3)34− decreased relative to that of single-charged ClO4−, and this effectively yielded more favorable ion-exchange competition by the ClO4−. Pilot-scale studies that used elevated nitrate levels in the water exhibited similar trends to the RSSCTs. These effects further emphasized the compounded competition that the uranyl carbonate posed on cetylpyridinium chloride-tailored GAC where this surfactant had been loaded in compact-micellar configuration. This represents an important consideration for a number of groundwater sources where uranium appears in trace concentrations.
Introduction
The removal of ClO4− from aqueous systems has been challenging as it persists for long times in aquifers. Perchlorate does not precipitate with cations (Horányi and Vértes, 1975; Brown and Gu, 2006) or sorb to the typical sediments minerals and soils to the extent that impedes its migration through aquifers (Urbansky and Brown, 2003; Brown and Gu, 2006). Also, ClO4− possesses low hydration energy and a small hydrated radius, and it thus has a remarkable affinity for ion exchange media (Gu and Brown, 2006).
A number of research teams have appraised the removal of ClO4− by ion exchange resins that include quaternary ammonium and pyridinium functional groups (Batista et al., 2000; Gu et al., 2001).
The approach used here has been to preload granular-activated carbon (GAC) with cationic surfactants that hosted quaternary ammonium or pyridinium functional groups, so that these functional groups could exchange the ClO4− (Parette and Cannon, 2005; Parette et al., 2005; Patterson, 2009). The positively charged heads of the surfactants are hydrophilic in nature and provide exchange sites for ClO4−. Pore size and volume of the GAC play a role in the formation of surfactant micelles, and thus affect the exchange capacities of the surfactant loaded GAC. The authors have noted that larger pore volume has facilitated more surfactant preloading, thus potentially availing more ion exchange sites (Parette et al., 2005; Patterson, 2009).
Selectivity is another important factor when designing ion exchange systems for ClO4−, since ClO4− is often present in lower concentrations than many of the other anions that it is competing with in the aqueous systems. The selectivity of media can be affected by the sorbate–sorbent interactions, the sorbate–solvent interactions (Xiong et al., 2007), and the functional groups and matrix. With respect to nitrate, Parette (2005) observed that the selectivity of ClO4−/NO3− was 33.6:1 for GAC that had been preloaded with cetylpyridinium chloride (CPC). This is compared to a selectivity of 10:1 for trimethyl ammonium ion exchange resins, as observed by Clifford and Weber (1983) and Gu et al. (2007).
Clifford (1999) characterized the selectivity sequence of relative affinities for the polystyrene trimethyl quaternary ammonium resin. In this analysis, ClO4−/Cl− relative affinity was 150:1, whereas UO2(CO3)34−/Cl− was 3,200:1, and the relative affinities for nitrate (3.2:1) and sulfate (9.1:1) were considerably lower.
A number of studies have addressed how the common ions nitrate (NO3−), sulfate (SO42−), and Cl− compete with ClO4− in pass-through granular media systems. Roach and Tush (2008) showed that the retention of ClO4− on the quaternary ammonium functional group decreased when chloride and SO42− were present in the poly (diallyldimethylammonium) chloride preparatory stock solution. Zhang et al. (2007) observed that the removal of ClO4− decreased in the presence of nitrate and other competing anions. They observed that NO3− significantly diminished the sorption of ClO4− on quaternary ammonium modified zeolite when nitrate concentrations were 2.5 to 10 times higher than that of ClO4−, and the ClO4− concentrations ranged from 100 to 400 mg/L. Najm et al. (1999) found that the efficiency of ion exchange systems to remove ClO4− decreased in the presence of SO42− and NO3−, when these competing anions concentrations were orders of magnitude greater than the ClO4− ion.
The competition that SO42− and NO3− pose with perchlorate is counter-balanced by the effect that ionic strength exerts on the species' activity. Specifically, the activity coefficients of divalent and multivalent ions decline more significantly than do those for monovalent anions. Thus, as noted by Clifford (1999), when a water's total dissolved solids (TDS) and ionic strength is increased, the medium removal of a monovalent anion can become preferred, relative to a divalent or multivalent ion, via the electroselectivity phenomenon. The uranyl carbonate system includes UO2(CO3)34− and UO2(CO3)22−, with a pKa of 7.76; these two species are by far the most prevalent ones that would appear in a water like that of Fontana, which contains some uranium and carbonate (see discussion below). As a yet further phenomenon, Clifford and co-workers have perceived that the UO2(CO3)34− species can actually be formed from the UO2(CO3)22− species within the ion exchange media, rendering uranium removal performance at a pH of 5.8 and above the pKa of 7.76 (Clifford and Zhang, 1994; Horng and Clifford, 1997).
When employing GAC media that have been preloaded with quaternary ammonium surfactants, the surfactants will array themselves as modified micelles, with the carbon walls affecting the configuration. Parette (2005) observed that when a 25%–30% loading of N-containing surfactant was preloaded into activated carbon, a mass and volume balance dictated that virtually all of the micropores, mesopores, and macropores were filled with the surfactant. Moreover, in pores that were wide enough (perhaps >30–50 A), the pores would be filled with rod-like or stacked-plate micelles. The micelles with charged heads outside and uncharged tails inside could configure into rods with 80 molecules around a circumference (Kalyanasundaram, 1987; Singh et al., 2005). The CPC molecules are 19.4 Å long, from tip of tail–to–N+ sites (Yei et al., 2005).
To the authors' knowledge, there have not been any refereed articles that characterized relative affinities when employing an activated carbon that has been preloaded with a pyridinium surfactant and also address how these anions compete with ClO4−. Thus, the aims of this study have been to appraise such tailored GAC media in regard to perchlorate's competition with NO3−, SO42−, Cl−, uranyl carbonate, and the other anions that are found in a native groundwater such as in Fontana, CA.
Experimental
Materials
All bench-scale and pilot-scale trials employed Aquacarb AC 2050AW carbon provided by Siemens Water Technologies. The Aquacarb AC 2050AW was an acid-washed anthracite-based GAC, initially of mesh 20 × 50 (0.85 × 0.3 mm). The # 20 × 50 mesh GAC was used in pilot-scale studies. For the Rapid Small-Scale Column tests (RSSCT) preparation, this anthracite GAC was ground and sieved to a US mesh size of 200 × 400 (75 × 38 μm). These carbons were preloaded with CPC, a cationic surfactant provided by Acros. The groundwater for this study originated from Fontana, CA. This groundwater contained the anion and metal species listed in Table 1.
No thiosulfate or other intermediate valence sulfur species were detected in Fontana groundwater.
U likely in form of uranyl carbonate; V, Cr, and Se likely present as the oxyanions vanadate, chromate, and selenate.
For the pilot study, the groundwater was pumped directly through to the on-site vessels. For the bench-scale study, this Fontana groundwater was shipped in 55-gallon drums to Penn State University, where the groundwater was stored, until use, under ambient conditions with incidental air contact through a cap that was not airtight.
Methods
Rapid small-scale tests
RSSCT employed the design and surfactant preloading protocol of Parette and Cannon (2005), based on proportional diffusivity. The columns were 0.5 cm in diameter and 13 cm in length, were filled with ∼1.27 g of GAC, and tailored with 33 bed volumes (BV) of 0.4% CPC surfactant solution in deionized water. The tailoring process took ∼2 days, during which surfactant solution was pumped and recirculated at 2 mL per minute through the activated carbon. This preloading protocol resulted in an RSSCT that contained 1.64 g of the CPC-loaded GAC (dry weight basis), which calculates to 29% loading of the CPC.
After the tailoring process, groundwater from Fontana was pumped into the tailored carbon at flow rates of 2.5 mL/min, which provided a 1 min empty bed contact time (EBCT). This simulated 11 min EBCT at the field scale level for a #20 × 50 mesh GAC based on proportional diffusivity.
Pilot-scale tests
The pilot-scale system consisted of two vessels in series (refer to Lutes et al., 2010 for details). Each contained 16.8 gallons (62 L) of surfactant-tailored GAC in the lead region (on top), and 8.4 gallons (32 L) of conventional GAC in the lower region (on bottom), which corresponds to a diameter of 14 inches (36 cm) and a height of 47 in (120 cm). All GAC used in the pilot columns was Aquacarb AC2050 AW (anthracite-based, acid washed). The conventional GAC was included to scavenge any surfactant that leached from the tailored media; during these runs, no surfactant passed through the conventional GAC. These pilot units operated at 1.5 gallons per minute average flows, which corresponded to an EBCT of 11.2 min as measured through the surfactant–tailored GAC in one vessel. For the control pilot system (which processed un-spiked native Fontana water), the flow rate was increased to 3 gallons/minute after the first vessel in series had exhibited perchlorate breakthrough, which corresponded to an 11.2 min EBCT, as measured through both of the vessel's surfactant tailored GAC media combined.
Stock solutions
For the RSSCT experiments, 10 g/L stock solutions provided the source of the spike of perchlorate to native Fontana groundwater. Stock solutions of sodium salts of NO3−, SO42−, and chloride were also used (refer to Patterson, 2009 for details). These salts were obtained from J.T. Baker. Table 2 shows all the experiments that were conducted and the concentrations of anions that were appraised for the RSSCT and pilot-scale experiments. Standards were prepared and refrigerated, and the standard concentrations were analyzed each time unknown anion analyses were conducted.
Listed numbers identify the total concentration of the species, after spiking.
Native nitrate in pilot-scale trials was 41 mg/L as NO3−.
Mean native total dissolved solids (TDS) in pilot-scale trails was 274 mg/L.
RSSCT, Rapid Small-Scale Column tests.
Anions and metals analysis
The common anions were analyzed with a Dionex DX-120 ion chromatography instrument, in accordance with the protocol by Patterson et al. (2010). This was equipped with an AS40 autosampler, 4 mm AS16 column, 4 mm AG16 guard column, 4 mm self-regenerating suppressor, and DS4 detection stabilizer. The stabilizer used a current of 300 mA and a temperature of 35°C. The elluent concentration employed was 25 mM NaOH. The sample loop for the ClO4− anion was 1,000 μL, whereas it was 25 μL for other anions.
The concentrations of the metals Cr, Se, U, B, P, V, Br, Mo, and Re were monitored by inductively coupled plasma mass spectrometry. These analyses were conducted using a Thermo Fisher Scientific X Series 2 inductively coupled plasma mass spectrometry at the Penn State Materials Characterization Laboratory.
Results
Rapid small-scale column tests
Nitrate competition
The RSSCT that used native Fontana water showed that ClO4− broke through to 1 μg/L at 19,000 BV (Fig. 1). This native Fontana groundwater contained 7 μg/L ClO4− and 32 mg/L nitrate as NO3−. This performance with media where the CPC had been loaded onto anthracite based GAC was not as favorable as in previous experiments for which CPC had been loaded onto a bituminous-based GAC that had more mesopores (Patterson, 2009).
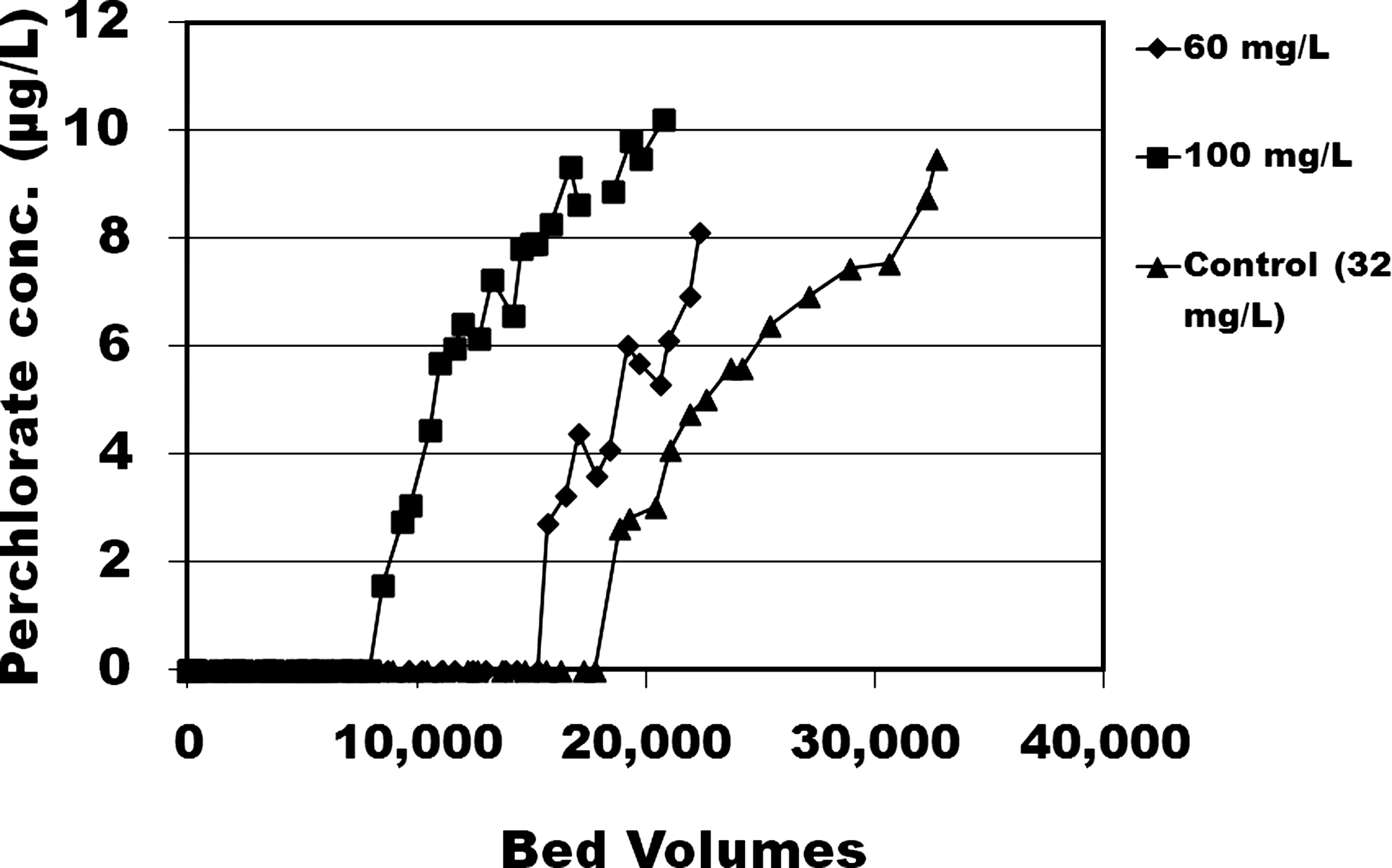
RSSCT experiments depicting the effects of ClO4− removal by CPC-tailored anthracite granular-activated carbon (GAC) when various concentrations of NO3− were spiked into Fontana groundwater. RSSCTs compared native Fontana groundwater (32 mg/L) to spiked total of 60 or 100 mg/L NO3−. CPC, cetylpyridinium chloride; RSSCT, Rapid Small-Scale Column tests.
In comparison to this 19,000 BV, when the Fontana water was spiked with nitrate to 60 mg/L total NO3−, the 1 μg/L ClO4− breakthrough occurred in 16,000 BV (16% less); with 100 mg/L total NO3−, the 1 μg/L ClO4− breakthrough occurred in 9,500 BV (50% less).
Thus, the higher NO3− concentration diminished the bed life for ClO4− removal. The authors note that with these concentrations of 30–100 mg/L NO3−, the NO3− itself has been shown to break through within 300–2,000 BV (Parette, 2005). Overall, these results reflect similar relative trends as have been observed for trimethyl ammonium resins media by Clifford (1999).
Chloride and bicarbonate competition
The effects of chloride and bicarbonate on ClO4− adsorption were also observed. When Fontana groundwater was spiked with Cl− to the secondary maximum contaminant level of 250 mg/L, ClO4− broke through to 1 μg/L at 25,000 BV (Fig. 2) and likewise with 550 mg/L HCO3−, the 1 μg/L ClO4− breakthrough occurred at 24,000 BV. The authors herein attribute this increased in bed life with increasing ionic strength to competition with UO2(CO3)3−4 as discussed below. Specifically, the increased ionic strength caused a considerably lower activity coefficient for this −4 charged U species.

Effects on ClO4− removal by CPC-tailored anthracite GAC when a high dose of Cl− and HCO3− was added to Fontana groundwater. RSSCTs compared native Fontana groundwater (12 and 200 mg/L) to that was spiked to a total of 250 mg/L Cl− and 550 mg/L HCO3−.
Sulfate and thiosulfate competition
When sulfate was spiked into the Fontana water, several factors played a role. With the 5 mg/L SO42−, the 1 μg/L ClO4− breakthrough occurred at 19,000 BV. However, when this water was spiked with 50 or 250 mg/L SO42−, the 1 μg/L ClO4− breakthrough occurred at 20,000 BV (Fig. 3). These results reflect two opposing trends: on the one hand, the higher SO42− concentrations corresponded to a higher TDS and ionic strength, which in the presence of UO2(CO3)3−4 species would diminish this U species' activity coefficient, and thus increase the ClO4− bed life in relative proportion to this (see discussion below). This effect was similar to that for Cl− and HCO3−. However, the increase in ClO4− bed life was not as great with the added SO42− competition as it was for the added Cl− and HCO3− above, and this was because the relative affinity of SO42− was stronger than that of the Cl− or HCO3−. These results indicate that when in the close packing micellar structure of CPC, within the GAC pores, the tightly knit pyridiniums posed some steric hindrance to the divalent SO42−, which with its high hydration energy and waters of hydration could not readily penetrate into this micelle structure. This steric hindrance was not exhibited for the monovalent NO3−, with its lower hydration energy (see discussion below).
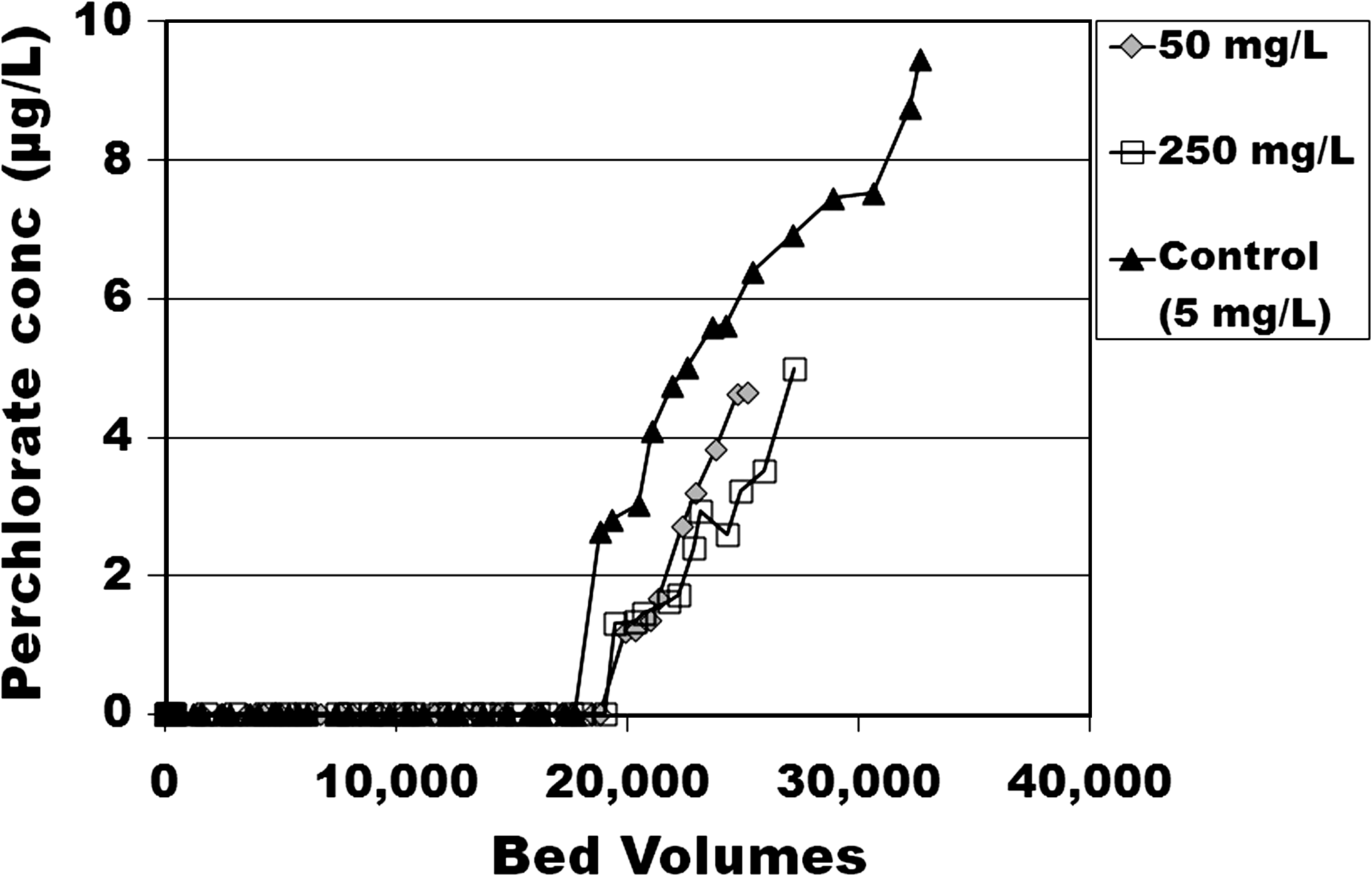
Effects on ClO4− removal by CPC-tailored anthracite GAC with various doses of SO42− are added to Fontana groundwater. RSSCTs compared native (5 mg/L) to spiked total (50 or 250 mg/L) SO42−.
Thiosulfate (S2O32−), an intermediate-valence sulfur species, was also spiked into the Fontana groundwater, at 1 mg/L and 10 mg/L concentrations, to appraise the effect of this oxyanion on the removal of ClO4− by the CPC-tailored carbon. The S2O32− had not been detected in the Fontana groundwater. Breakthrough at 1 μg/L ClO4− occurred slightly sooner, with a 12% decrease in ClO4− bed life when competing with 1 mg/L S2O32− and a 20% bed loss with 10 mg/L S2O32−. These trends reflected that S2O32− indeed competed with ClO4−, but there was some size exclusion steric hindrance posed for the −2 charged S2O32− in the presence of the surfactant pyridinium micelles, just as there was for the −2 charged SO42− (Patterson, 2009; Patterson et al., 2010).
The authors had earlier observed considerably more S2O32− competition when dicocoalkyldimethylammonium chloride (Arquad 2C-75) was used as the preloading surfactant and the RSSCT-processed Redlands, CA, water (Patterson, 2009; Patterson et al., 2010). In that case, the trimethylammonium surfactant apparently did not exhibit as significant a steric hindrance.
Pilot-scale tests
In conjunction with column tests, pilot-scale experiments were also conducted at Fontana, CA. These pilot studies also aimed to appraise the relative anion competition. The pilot studies employed two beds in series. Each bed hosted CPC-tailored anthracite GAC in the top two-thirds of the bed, and conventional anthracite GAC in the bed's bottom third. Results from the pilot scale showed that NO3− affected the adsorption of ClO4− to surfactant-tailored GAC. Perchlorate was analyzed as water exited the first stratified bed (Effluent 1) and also the second stratified bed (Effluent 2). For Fontana groundwater, 1 μg/L ClO4− broke through from the first bed at 12,000 BV, where BV were measured through the first column's CPC tailored GAC media (Fig. 4). Likewise 1 μg/L ClO4− broke through the second bed in series at 12,000 BV, where BV were as measured through the two columns of CPC tailored GAC media together (Fig. 5).

Effluent from the first pilot bed in series: Effects on ClO4− removal via CPC-tailored anthracite GAC when various concentrations of NO3− and SO42− were used. Shown are the total concentrations of NO3− (60 mg/L) and SO42− (444 mg/L) in each experiment. Control had a background concentration of 32 mg/L NO3− as NO3− and 5 mg/L SO42−.
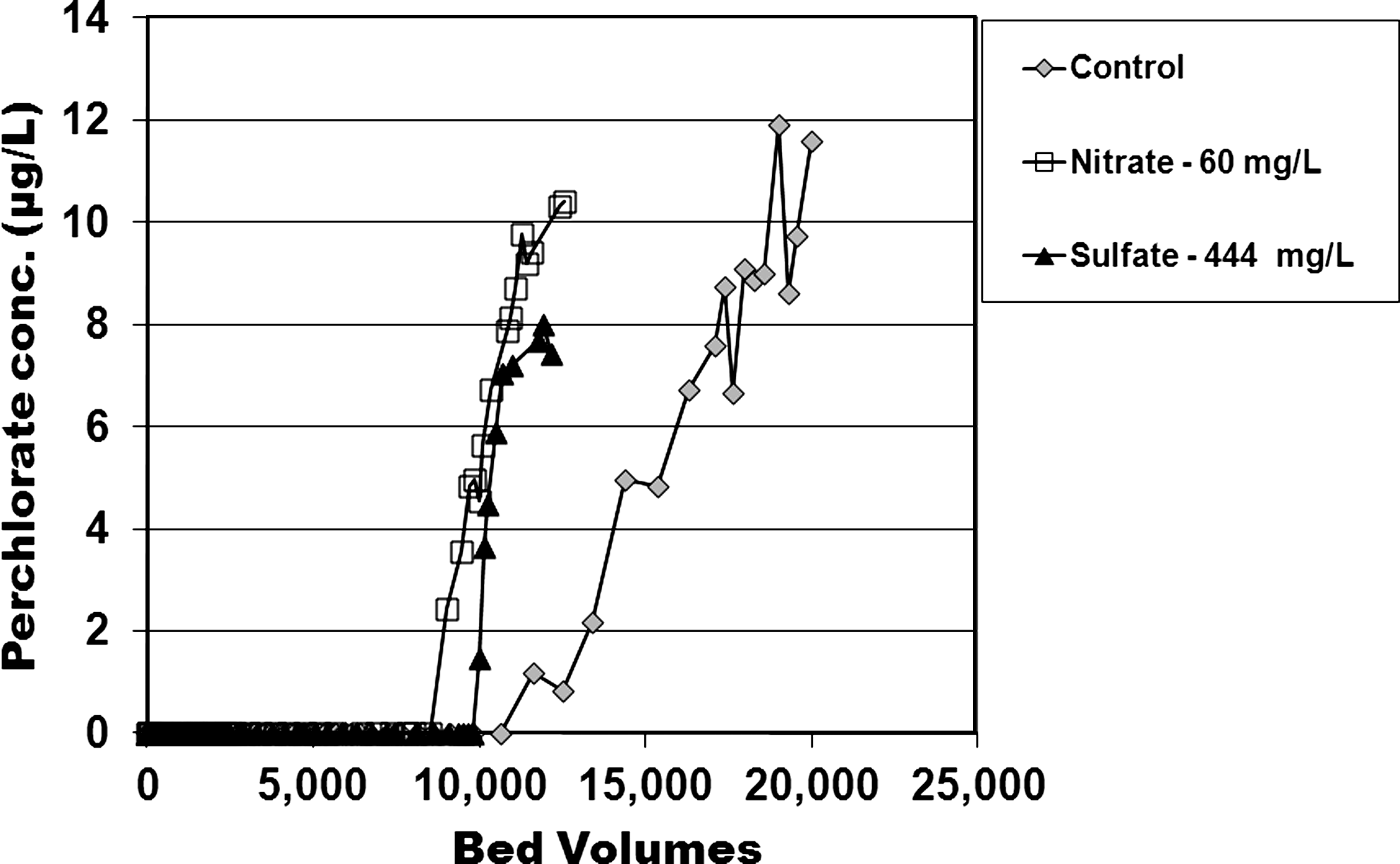
Effluent from the second pilot bed in series: Effects on ClO4− adsorption to CPC-tailored anthracite GAC when various concentrations of NO3− and SO42− were used. Shown are the total concentrations of NO3− (60 mg/L) and SO42− (444 mg/L) in each experiment. Control had a background concentration of 32 mg/L NO3− as NO3− and 5 mg/L SO42−.
The relative influence of increased NO3− in the pilot units was consistent with that observed in the RSSCTs above, namely, 60 mg/L NO3− diminished bed life by about 20%–25%. The diminishing effect was somewhat greater through the second bed than through the first.
A total of 444 mg/L SO42− posed about the same competition as did 60 mg/L NO3− (Figs. 4 and 5). The fact that it took such a high SO42− dose to exhibit the same competitive effect as 60 mg/L NO3− again highlighted that the CPC micelle structure posed steric hindrances to the divalent sulfate species.
Competition and presence of other oxyanions in fontana water
For the pilot columns that processed 60 mg/L NO3−, the authors also monitored key effluent samples for nine metals, etc., that can form oxyanions as presented in Table 3. As shown, the Fontana water notably contained 1.9 μg/L uranium (U), 8 μg/L vanadium (V), and 3.3 μg/L chromium (Cr). The uranium thermodynamics at pH 7.5 in an abundantly alkaline water dictates its presence as UO2(CO3)32− or UO2(CO3)34− (pKa 7.75), with the latter predominating within the media (Zhang and Clifford, 1994; Clifford and Zhang, 1994; Horng and Clifford, 1997). The V would appear as the oxyanion vanadate (H2VO4−) (Greenwood and Earnshaw, 1997), and Cr as chromate (HCrO4−). As a passing note, one observes that the B and P effluents at 58 BV were higher than at other times. This reflected roll-over of the phosphate and borate oxyanions, and the CPC surfactant offered only limited exchange capacity for these two species.
Concentrations reported as μg of the metal/L.
Beds volumes based on volume of tailored media in Beds 1 and 2 combined.
<DL, below detection limit.
Intriguingly, for this Fontana groundwater, the breakthroughs of ClO4−, U species, and V species all occurred at the same 9,500 to 10,000 BV (Fig. 6). As noted in Clifford, 1999 for ion exchange media, the relative affinity for UO2(CO3)34− is more than an order of magnitude greater than ClO4−. In this light, the Fig. 6 behavior can be explained as the UO2(CO3)34− anion displacing both the ClO4− and the H2VO4− as the mass transfer zone of the UO2(CO3)34− progressively moved through the CPC-GAC media. This perspective is emphasized by noting that both the ClO4− and H2VO4− exhibited rollover, with effluent concentrations considerably exceeding the influent levels of 7 μg/L ClO4− and 8 μg/L V. Nearly two-thirds of the ClO4− that had adsorbed initially was subsequently desorbed. This result highlights the strong affinity of the pyridinium for the UO2(CO3)34− anion.
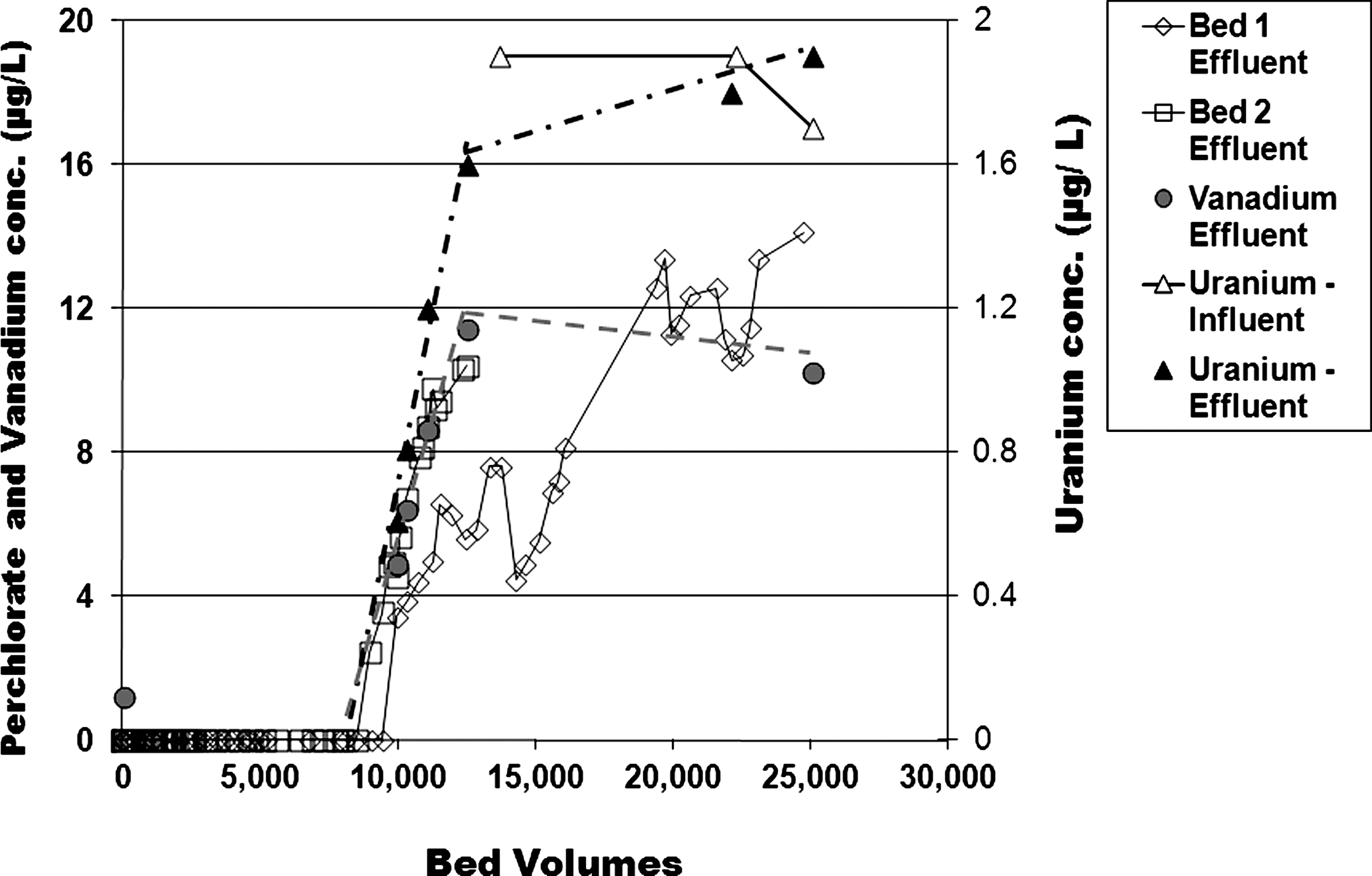
Effluent from first and second pilot bed series: When spiking to 60 mg/L total NO3−, effluent concentration of ClO4−, U, and V.
The observation that both the perchlorate and vanadate exhibited breakthrough when the uranyl carbonate species broke through may be counter intuitive to conventional ion exchange behavior, when exchanging commonplace ions that all have relative affinities that are within a factor of 2–5 of one another (e.g., 3.2 for nitrate vs. 9.1 for sulfate—refer to Clifford, 1999). In contrast, the Fontana water contained perchlorate with its 150:1 relative affinity that is more than an order of magnitude higher than the common ions. It also contained UO2(CO3)34−, whose relative affinity of 3,200 is yet another order of magnitude greater than for perchlorate. Thus, significantly, by the time 10,000 BV of Fontana water had been processed, the media's perchlorate capacity was far from exhausted; that is, the perchlorate mass transfer zone was well above the bottom of the bed. Nonetheless, as the uranyl carbonate exchanged to the media, it displaced perchlorate, vanadate, and anything else. Also, the uranyl carbonate, with its −4 charge, could concurrently strongly bond to four quaternary ammonium functional groups. This could disrupt the micellar packing of the surfactants, and effectively block further internal access to the perchlorate, vanadate, or even further uranyl carbonate. Other additional empirical evidence for this disruption is explored by Patterson (2009).
The disruptive impact of the UO2(CO3)34− is further emphasized by Fig. 7 pilot-scale data, which compares the ClO4− bed life when processing Fontana water and the Fontana groundwater spiked to 315 μg/L ClO4− (mean value). While this ClO4− dose was 40 times higher than the native, the bed life to a given proportion of breakthrough was a mere 35%–40% less than for the native Fontana water that contained 7 μg/L ClO4−. Thus, ClO4− performance was not dictated so much by the number of active pyridinium sites that could be occupied by the ClO4− as by the intense displacement of the sorbed ClO4− by the disrupting UO2(CO3)34− anion. For this surfactant-tailored media, the ion exchange sites are arranged around the periphery of compact micelles, and the individual surfactant molecules could be construed as more pliable than sites that are attached into a conventional ion exchange polymeric backbone. The competition with uranyl carbonate was likely exaggerated by this surfactant pliability, and the micelle structure could have been disrupted by this strong bonding with the uranyl carbonate.
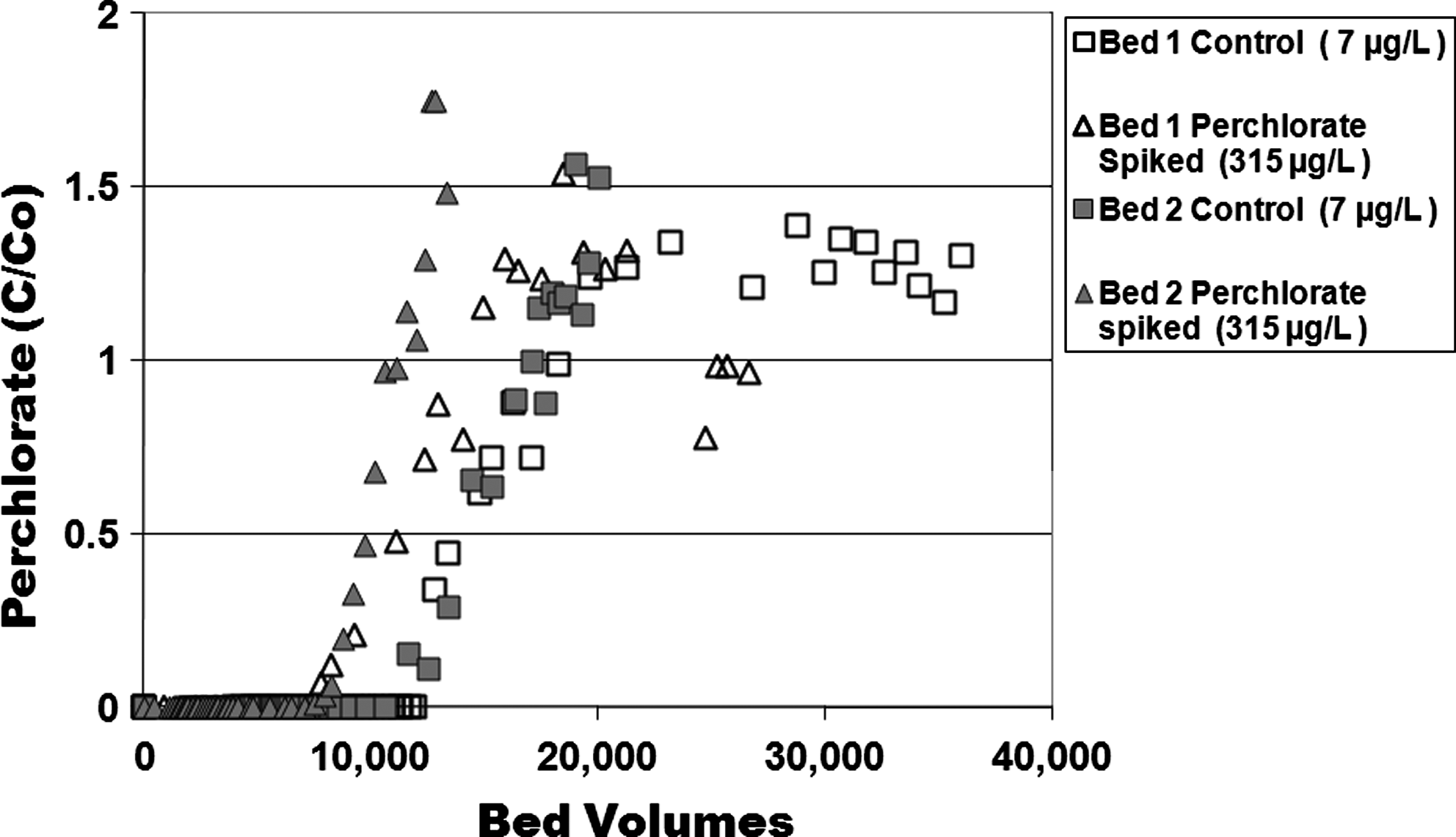
Effluent from first and second pilot bed in series: Comparison to spiking native Fontana water to 315 μg/L perchlorate (mean value).
Discussion
Uranyl cabonate competition, and activity coefficients
The data have shown that an increase in HCO3−, Cl−, or SO42− caused an increase in ClO4− bed life. This increased ClO4− bed life is explained by considering the ion exchange competition with the uranyl tricarbonate species (UO2 (CO3)34−), particularly relative to its activity coefficients. This Fontana water contained ample HCO3−, and at the ambient pH 7.5, one of the predominant forms of U dissolved in such an alkaline water is the UO2(CO3)34− species (as confirmed via modeling by the authors herein for this Fontana groundwater via Visual MINTEQ 3.0) (Jang et al., 2007). Moreover, when uranyl carbonate is present at pH 7.5–8, it is the UO2(CO3)34− species that effectively is formed on the ion exchange media (Clifford, 1999).
The ion exchange equilibrium expression that relates ClO4− to UO2 (CO3)34− would take the form of Equation 1 (adapted from Clifford, 1999).
where K is a separation constant, qu and qc are the media-phase concentrations of the UO2(CO3)34− and ClO4− respectively, γ−1 and γ−4 are the aqueous activity coefficients of the ClO4− and UO2(CO3)34− respectively, and the [] denote aqueous concentration (eq/L water). The BV to ClO4− breakthrough are related to (but not equivalent to) the media-phase concentrations. Using the Debye–Hückel approximation (Snoeyink and Jenkins, 1980), the γ−1 of the native Fontana groundwater is computed as 0.92, whereas γ−4 is computed as 0.28, and (γ−1)4/γ−4 is 2.6. In comparison, for the Fontana groundwaters that were spiked to either 250 mg/L SO42−, or 250 mg/L Cl− or 500 mg/L HCO3−, the TDS was increased to 640–660 mg/L and the γ−1 was 0.88, γ−4 was 0.14, and (γ−1)4/γ−4 was 4.2 (i.e., 60% more than for native water). It stands to reason that as these activity coefficient ratios increased, the media concentration of ClO4− would increase relative to the media concentration of the uranyl carbonate species. Further, this would correspond to increasing bed life for ClO4− removal. It is these trends that are depicted by the data in Figs. 2 and 4. When TDS and ionic strength increased, and the ratio (γ−1)4/γ−4 increased by 60%, the bed life increased by 10%–32%. The greatest increase in bed life was manifest for the Cl− and HCO3− anions, and it was these anions that posed the least competition selectively, relative to ClO4−.
For the pilot units, where the SO42− level increased to 444 mg/L and total TDS was 810 mg/L, the γ−1 was computed as 0.87, γ−4 was 0.12, and (γ−1)4/γ−4 was 4.7. On this basis of the ClO4− competing with UO2 (CO3)34−, the ClO4− bed life could yet further increase with the diminished γ−4. However, at this high SO42− dose, the SO42− also competed with the ClO4−, which would diminish bed life, and the net effect was a balance of these two phenomena.
Steric hindrance of surfactant pyridinium micelles
For the surfactant pyridinium micelles herein, NO3− posed greater competition to ClO4− than did SO42−. This behavior was somewhat distinct from the behavior of conventional methyl quaternary ammonium ion exchange media where SO42− has been the greater competitor (Smith and Woodburn, 1978; Clifford and Weber, 1983). The reasons for this distinction relate to the size exclusion effects incurred by the rod-like or stacked-plate micelles that the CPC formed into. As discussed above, CPC can configure into rod-like micelles with 80 molecules around a circumference, and a 38.8 Å diameter. This translates to about 1.5 Å from the center of one N+ in a pyridinium group to the N+ in the adjacent pyridinium group, when the flat sides of the pyridinium groups are aligning with one another (or about 4–5 Å in edge-to-edge alignment).
Significantly therefore, the pyridinium functional groups in these micelles are more tightly and uniformly spaced than are the functional groups in resin-based ion exchange media. This configuration impacts both the access of anions to the N+ site through the ring structure periphery of the pyridinium group, and also alignment of the anions with this N+ site. It is this alignment that apparently causes the steric hindrance on SO42− exchange.
Hydration energy may also pose a role, as Sata et al. (1997) and Behnsen and Riebe (2008) observed that anions with low hydration energy were more selectively exchanged than anions with high hydration energy. Anions with higher hydration energy associate with more waters of hydration, and thus behave as if they are larger species. Further, ClO4− hosts the lowest ΔG of −214 kJ/mol. Next, NO3− and Cl− are about the same as one another (306–347 kJ/mol), and SO42− is the highest at 1,000 kJ/mol (Marcus, 1985; Sata et al., 1997). In particular, relative to the micellar structure herein, this trend would be emphasized in that SO42−, with its larger number of waters of hydration, could not penetrate the pyridinium periphery as readily as did NO3− and ClO4−. Also, with the tight micelle structure, the SO42− will encounter steric hindrances when concurrently exchanging at two N+ sites.
Conclusions
The present study shows that for this natural Fontana, CA, water, anions such as nitrate and thiosulfate competed with ClO4− for adsorption sites, whereas sulfate did not compete until its concentration was very high. Uranyl carbonate competition posed a significant limitation to perchlorate removal capacity for GAC that was preloaded with CPC surfactant. When the Fontana water was spiked with higher levels of chloride or bicarbonate, the BV to breakthrough of perchlorate actually increased. This was linked to the consequently diminished activity of the competing uranyl carbonate species, with its −4 charge, as caused by the higher ionic strength incurred by the chloride or bicarbonate. These effects further emphasized the compounded competition that the uranyl carbonate posed on CPC-tailored GAC where this surfactant had been loaded in compact-micellar configuration. This represents an important consideration for a number of groundwater sources where uranium appears in trace concentrations.
Footnotes
Acknowledgments
This research was funded in part by Environmental Security Technology Certification Program (ESTCP) Project ER-0546 Contract W 912HQ-06-C-005, and by National Science Foundation contract 0829092. Thanks are extended to the City of Fontana for providing the groundwater used in these tests. The authors would also like to thank Dr. Waleska Castro of the Materials Characterization Laboratory at the Earth and Environmental Systems Institute at Pennsylvania State University for conducting the metals analysis. The authors also thank James Graham and his team from Siemens Water Technologies for preparing the pilot-scale systems and the CPC-tailored anthracite GAC.
Author Disclosure Statement
No competing financial interests exist.