
Abstract
Abstract
Release of metals from fly ash stored in ash impoundments can pose environmental risks. Laboratory-scale impoundments loaded with Class C and Class F fly ashes were studied to investigate the release of metals during aging, changes in the mineralogy of the ash, and associated changes in the leachability of the metals. The water column above the settled ash was sampled over 9–12 months, and cores of ash were regularly collected from one of the Class C ash impoundments and subjected to a sequential extraction procedure that targeted metals bound by specific mechanisms. The long-term nature of experiments and integration of aqueous- and solid-phase analyses are unique aspects of the study that yielded insights into the fate of metals during fly ash storage and disposal. For Class C ashes, high concentration of alkaline solids led to pH values as high as 11.6 that gradually declined as the impoundment took up carbon dioxide from the atmosphere. A considerable fraction of the selenium in the ash (∼50%) was rapidly released to solution, and measurable release of chromium (>0.7%) to the water column also occurred quickly. Slower release of arsenic was observed. For the Class C ash impoundment with sampled cores, the total concentrations of arsenic and chromium did not change substantially, but these elements became more resistant to chemical extraction with time. The high pH maintained in the ash resulted in the formation of solid phases that may reduce metals release.
Introduction
Ash impoundment releases are an environmental concern due to the potential leaching of toxic metals and metalloids from the ash to the water and the subsequent discharge of waters from the impoundments. Numerous studies have examined the leaching potential of Se and As from fly ash (Huggins et al., 2007; Jegadeesan et al., 2008; Khodadoust et al., 2011). Metal mobility is controlled by the dissolution of primary solids and precipitation/sorption reactions. Class C fly ash has a high calcium content and produces alkaline fluid conditions, which would result in minimal adsorption of arsenic and selenium to the fly ash (Rowe et al., 2002; Al-Abed et al., 2007; Wang et al., 2009). Chemical characterization of fly ash can be determined using sequential extraction procedures. Such experiments have shown that arsenic and selenium leaching from alkaline fly ash was controlled by a calcium-containing phase (Rowe et al., 2002; Kim, 2006).
The objective of this study was to examine the mobilization of metals from the settled ash layers of impoundments that store fly ash. A further objective was to probe the relationship among the evolution of the fly ash mineralogy, trace element speciation, and leachability during aging. Long-term laboratory-scale ash impoundment studies were performed using Class C and Class F ashes from power plants in the United States and India. The study focused on selenium, arsenic, and chromium. For the U.S. Class C ash, a detailed examination of the changes in mineralogy and metal speciation was conducted through the integration of X-ray diffraction (XRD) analyses and sequential extractions.
Experimental Protocols
Materials
Experimental ash impoundments were designed to explore metal leaching from fly ash in poorly mixed environments. The ashes were mixed with water at a 1:1 ratio for Class C ashes and 2:1 for Class F ashes by mass, and then added to 20-L plastic containers to create impoundments with an ash depth of 20 cm. This ratio resulted in ash layers that could be sampled at specific depths as well as a free water column of 10 cm above the ash that could be easily maintained by weekly addition of water to compensate for evaporation. The impoundments were open to the atmosphere. One impoundment used a mixture of ultrapure water and Class C ash from combustion of Powder River Basin sub-bituminous coal at a U.S. power plant (Table 1) that was characterized in detail (as “Ash B”) in a previous study (Luo et al., 2011).The ash has a high CaO content (∼19% by mass) and contains trace amounts of lime in addition to silicate and aluminosilicate minerals. The ash has a low unburned carbon content (0.32% by mass) and a specific surface area of 1.4 m2/g. It consists primarily of spherical particles with sizes of 0.1–10 μm. Three other impoundments used fly ash materials from Indian power plants with similar particle sizes prepared with rainwater collected in Mumbai, India. Indian Ash 1 was a Class C ash, and Indian Ashes 2 and 3 were Class F ashes. The initial pH values of the ultrapure water and rainwater were 5.6 and 5.9, respectively, and the rainwater had very low dissolved solids compared to the composition following contact with ash.
Ash impoundment sampling
Free water column samples were collected 2.5 cm above the surface of the settled ash with 10-mL syringes and filtered with 0.2-μm polytetrafluoroethylene filters. Cores of ash (20-cm length) at aging times from 1 to 36 weeks were obtained from the U.S. ash impoundment using a 0.64-cm inner diameter copper tube. The ash was collected in aluminum drying pans and separated into three equal-length sections by depth. Each ash sample was weighed, and then air-dried for 1 week at room temperature. A small mass (∼0.3 g) from each sample was oven-dried at 105°C and reweighed to determine the solids content. The height of the ash–water interfaces did not change over time, although from coring, it is clear that the ash is more densely packed at a greater depth.
Sequential extraction
A five-step sequential extraction scheme based on one developed by Tessier et al. (1979) was applied to the ash materials recovered from the U.S. ash impoundment (Table 2). Each step targeted metals bound by different mechanisms or associated with different phases. The sequential extraction used 500 mg of ash on a dry basis. The solid to liquid ratio was maintained at 10 mg/mL. Following each step, the samples were centrifuged at 15,000 g (relative centrifugal force), and the supernatant was collected.
Analytical methods
Trace metal concentrations were measured by inductively coupled plasma (ICP) mass spectrometry for the U.S. ash (Agilent 7500ce) and ICP optical emission spectrometry for the Indian ashes (Jobin Yvon Horiba Ultima 2000). Calibration standards were prepared using the extraction fluids to avoid potential matrix effects, and analytical runs included frequent check standards and blanks for quality control. The recoveries of trace elements were examined by application of the extraction procedure to the National Institute of Standards and Technology reference material for elements in coal fly ash (Standard Reference Material® 1633b), and total digestions yielded concentrations within 20% of the certified values for arsenic and selenium. XRD was used to examine the evolution of the ash mineralogy in the U.S. ash impoundment over time. XRD was performed on a Rigaku D-MAX/A diffractometer that uses Cu-Kα radiation and has a vertical goniometer and a scintillation counter.
Results
Water sampling
The free water column compositions were monitored over time to track the release of elements from the ash (Figs. 1 and 2). Results are shown for the three trace elements and for major elements that displayed interesting temporal trends. The concentration of dissolved calcium in the U.S. ash impoundment increased rapidly, and then declined dramatically between weeks 3 and 10. The pH initially increased to 11.04 due to the dissolution of CaO, and then decreased as the water took up atmospheric CO2 and CaCO3 precipitated. The Indian Class C ash impoundment also had high initial pH and dissolved calcium concentrations that declined as the water took up CO2.
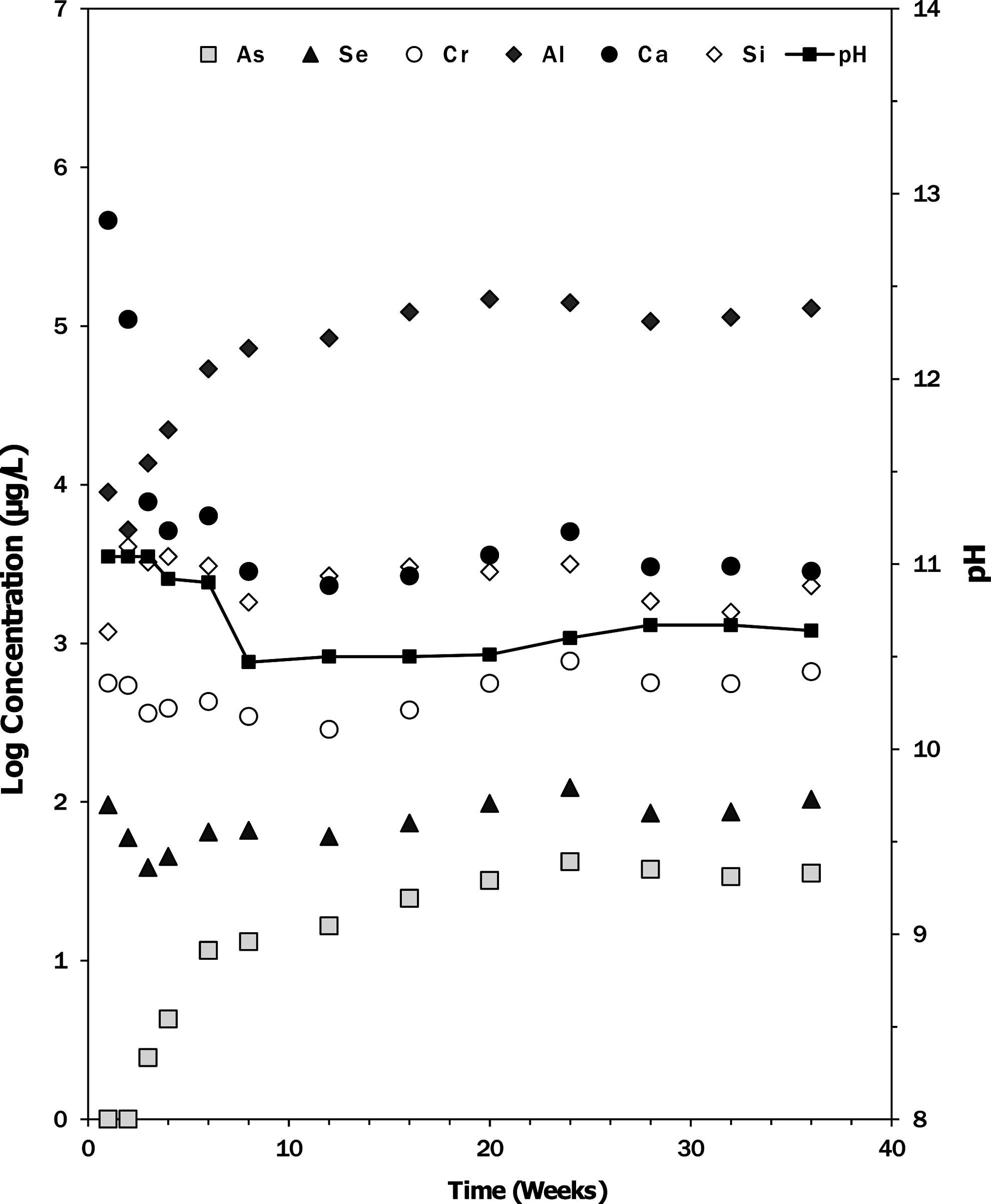
Dissolved concentrations of major and minor elements (μg/L) in the water column of the U.S. Class C fly ash impoundment as a function of time (number of weeks since impoundment formation) as determined by inductively coupled plasma mass spectrometry. pH of the water phase is shown on the secondary axis.
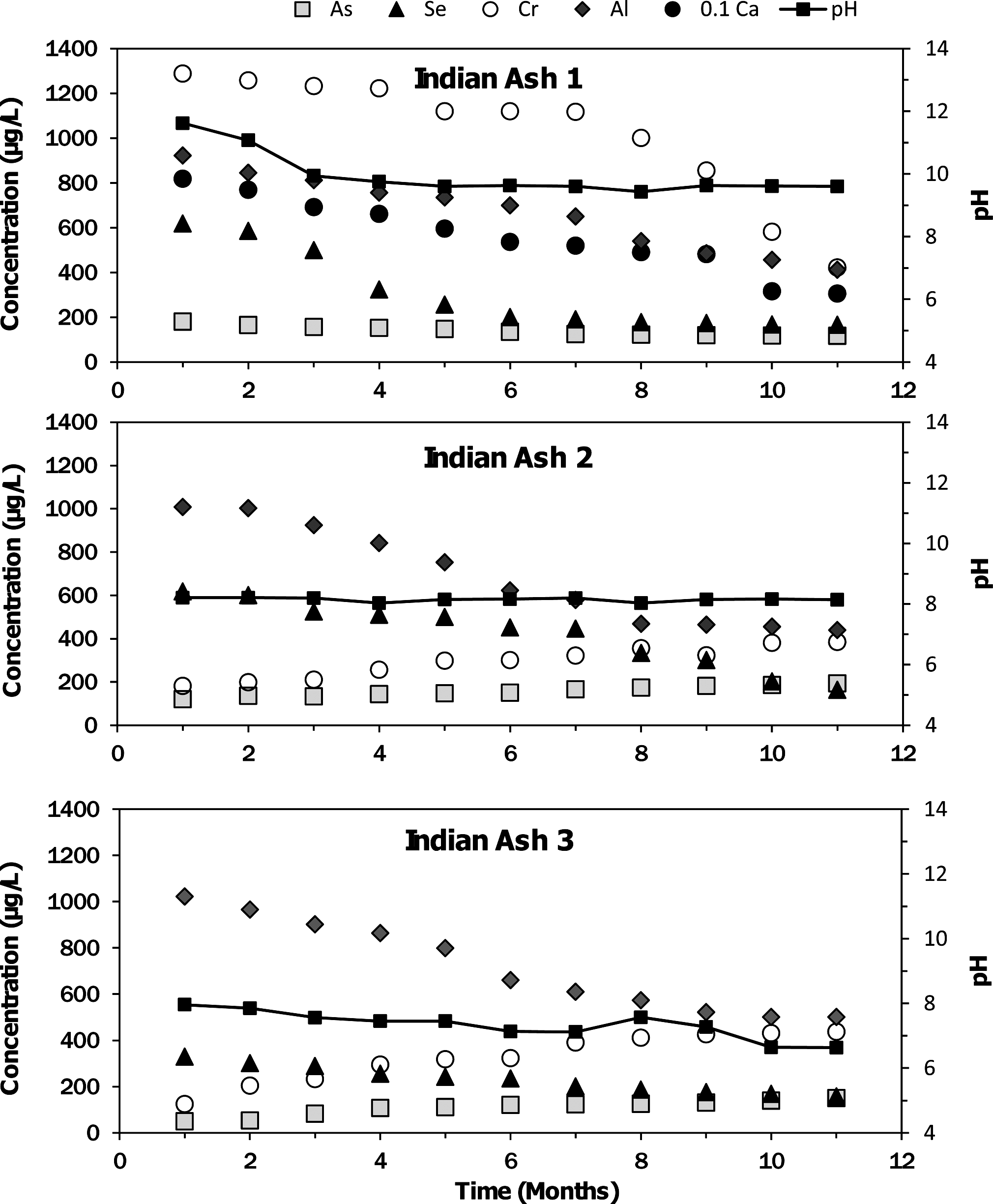
Dissolved concentration of elements (μg/L) in the water column of Indian fly ash impoundments as a function of time (number of weeks since impoundment formation) as determined by inductively coupled plasma optical emission spectrometry. pH of the water is shown on the secondary axis.
The concentrations of dissolved chromium and selenium in the U.S. ash impoundment exhibited similar trends to the dissolved calcium. The measured concentrations of both minor elements were high during the first week (Se=96 μg/L, Cr=561 μg/L), decreased through week 3 (Se=39 μg/L, Cr=362 μg/L), and then increased back to the initial concentration observed during week 1 (Fig. 1). The Indian Class C ash impoundment had high initial concentrations of chromium and selenium that both decreased over time (Fig. 2).
The concentrations of aluminum and arsenic in the water column of the U.S. ash impoundment increased continuously before plateauing (Al=140,000 μg/L, As=42 μg/L) after 24 weeks (Fig. 1). As the pH decreased, the concentrations of As and Al increased. The arsenic concentration in the Indian Class C ash impoundment also had a slow increase from 120 to 193 μg/L over 12 months.
The two Indian Class F ash impoundments had much lower initial pH values (8.2 and 8.0) than the Class C ash impoundments. The pH of the impoundment with Ash 2 was constant, but the pH of the impoundment with Indian Ash 3 decreased during the experiment to 6.6 (Fig. 2). Both ash impoundments had high initial selenium concentrations that decreased throughout the duration of the experiment, low initial chromium concentrations that increased with time, and arsenic concentrations that also increased with time.
Sequential extraction
The sequential extraction of the U.S. ash impoundment cores did not show significant variations in major element compositions with depth or time (Fig. 3). The concentrations of selenium, aluminum, and calcium in the ash layer did not change after the initial impoundment formation, but compared with the original ash about half of the selenium was released during the initial formation (Fig. 4). Although the total recoveries of arsenic and chromium were unchanged with time, the fraction extracted in the dithionite-citrate-bicarbonate (DCB) step decreased with time, while that extracted in the heated acid digestion increased through week 16 (Fig. 5), although the trend slightly reversed to week 20. This trend suggests that arsenic and chromium in the ash were transformed into more recalcitrant solid phases.
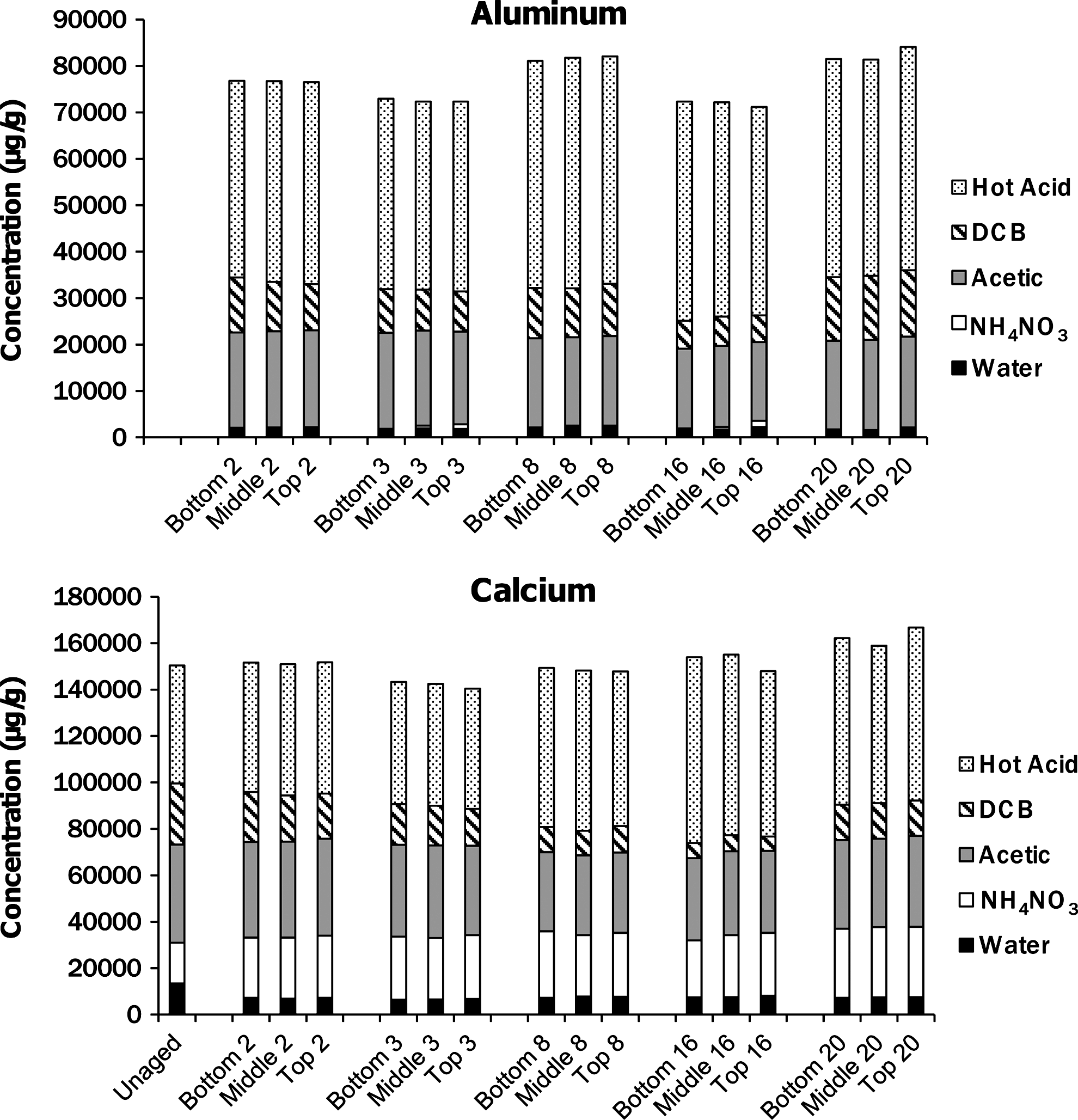
Major element recovery (μg/g ash) from each step of sequential extraction of the U.S. Class C fly ash as a function of time (weeks since impoundment formation) and depth within the ash core. The x-axis labels indicate the depth from which the ash was collected (top is the location closest to the ash–water interface) and the number is the weeks since the formation of the impoundment. Fractions mobilized in water and ammonium nitrate are the bottom two sections of each column, and in some cases the amounts were so small that they do not appear as part of the column. Sequential extraction results for aluminum for the unaged ash were not available.
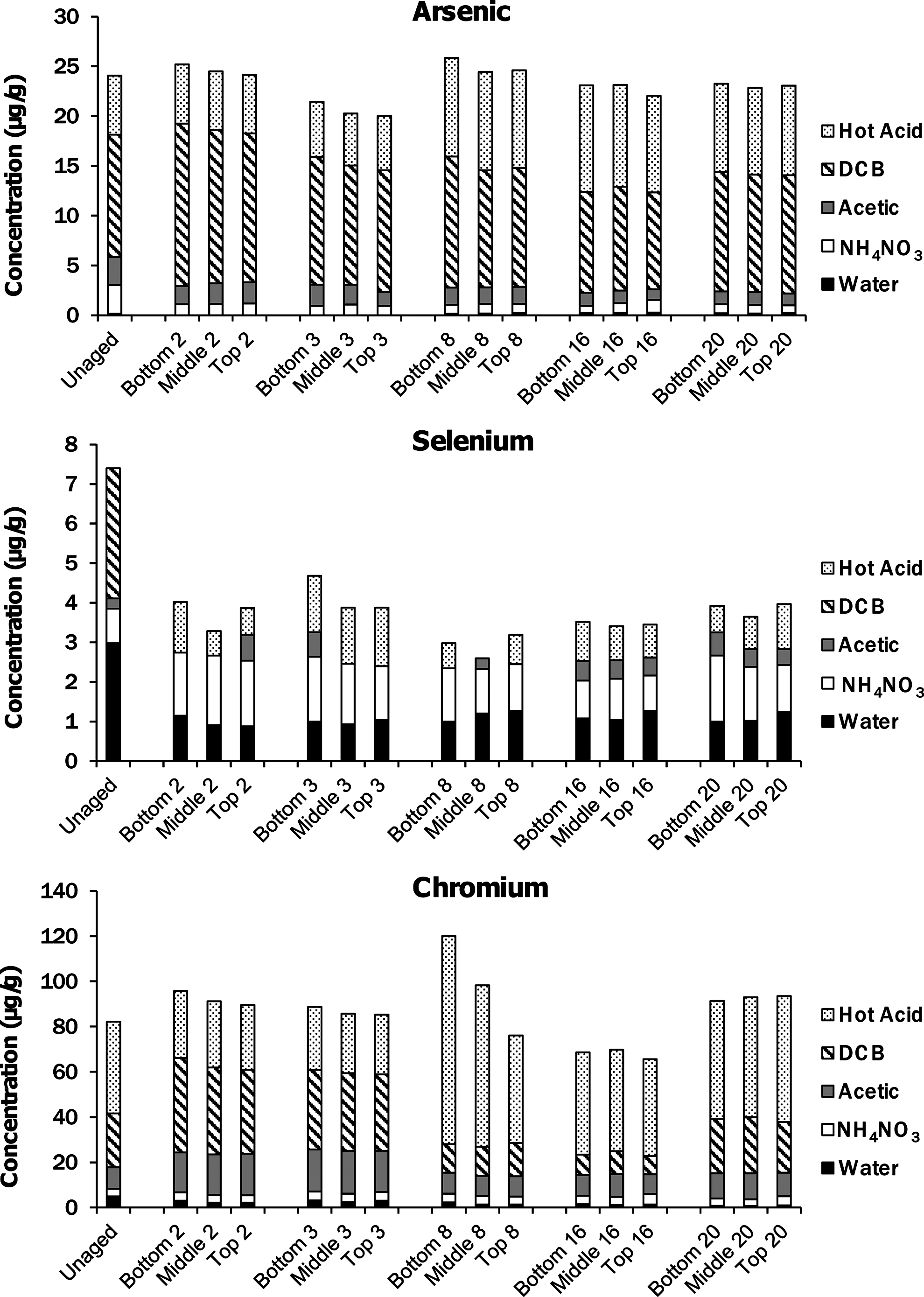
Minor element recovery (μg/g ash) from each step of sequential extraction of the U.S. Class C fly ash as a function of time (weeks since impoundment formation) and depth within the ash core. The x-axis labels indicate the depth from which the ash was collected (top is the location closest to the ash–water interface) and the number is the weeks since the formation of the impoundment. Fractions mobilized in water and ammonium nitrate are the bottom two sections of each column, and in some cases, the amounts were so small that they do not appear as part of the column.
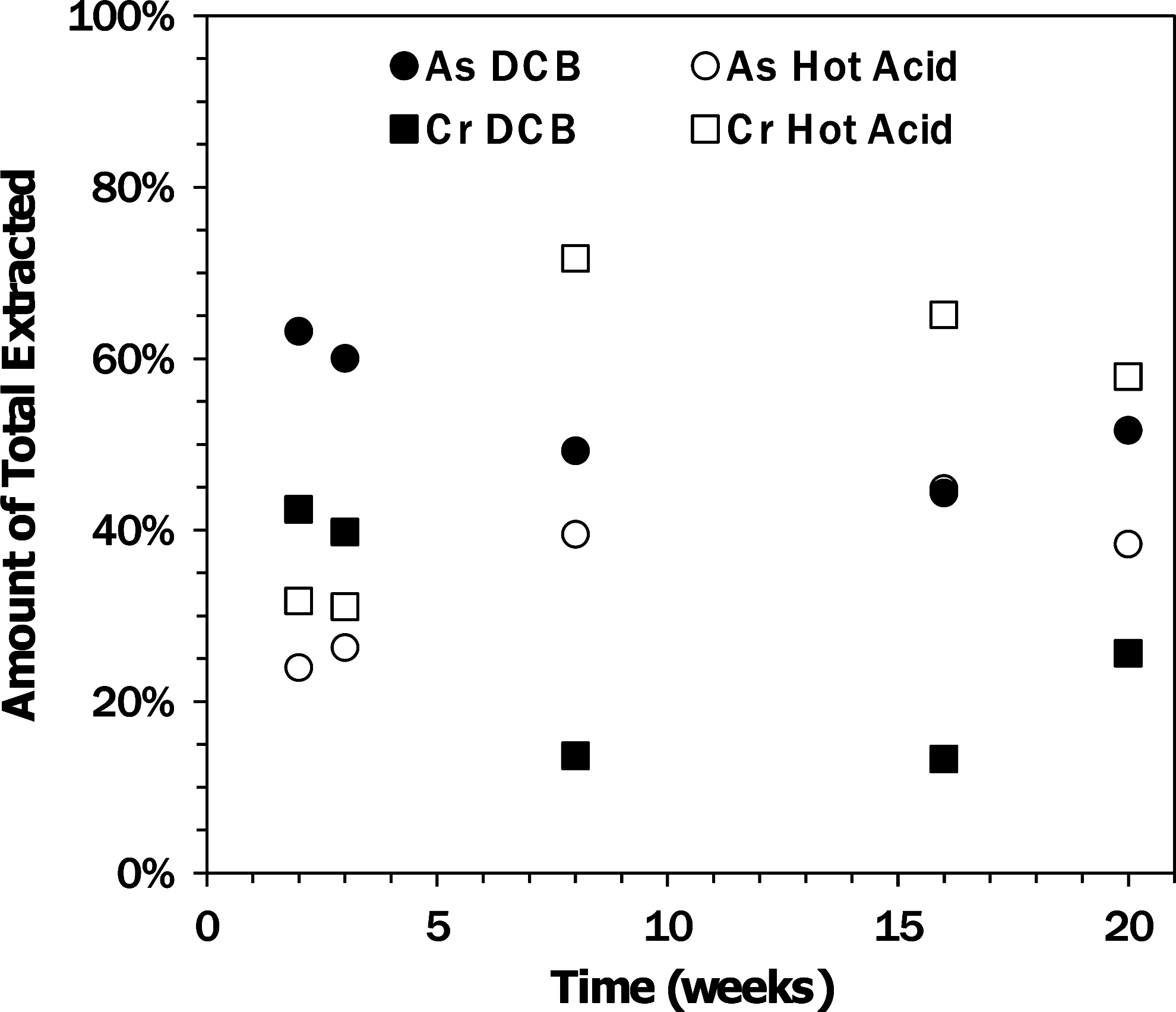
Relative amounts of total mass of arsenic and chromium extracted in the extraction steps with dithionite-citrate-bicarbonate (DCB) and concentrated nitric and hydrochloric acids at 100°C (hot acid). The arsenic data for 16 weeks for both DCB and hot acid are overlapping.
X-ray diffraction
The XRD pattern of the unreacted U.S. Class C ash was dominated by quartz (SiO2) with minor amounts of anhydrite (CaSO4), gehlenite (Ca2Al[AlSiO7]), merwinite (Ca3Mg[SiO4]2), periclase (MgO), and aluminosilicate glasses. The XRD patterns of ash aged in the impoundment indicated the formation of ettringite (Ca6Al2[SO4]3[OH]12·26H2O), vertumnite (Ca8Al4[Al4Si5]O12[(OH)36·10H2O]), and calcite (CaCO3) over time. The formation of calcite is consistent with the uptake of CO2 from the atmosphere into the high pH and high calcium solution of the water column. The formation of calcite can also be tracked by the declining calcium concentrations in the water column. Ettringite was also observed as a fly ash weathering product in previous studies (Zevenbergen et al., 1999). Vertumnite is a zeolite that could potentially adsorb trace elements released from other phases in the ash. The formation of both ettringite and vertumnite were probably promoted by the ash layer environment of high pH with solutions rich in silicon and aluminum. Anhydrite (CaSO4) was present in the initial unreacted ash, but it rapidly dissolved in the ash impoundment (Fig. 6).
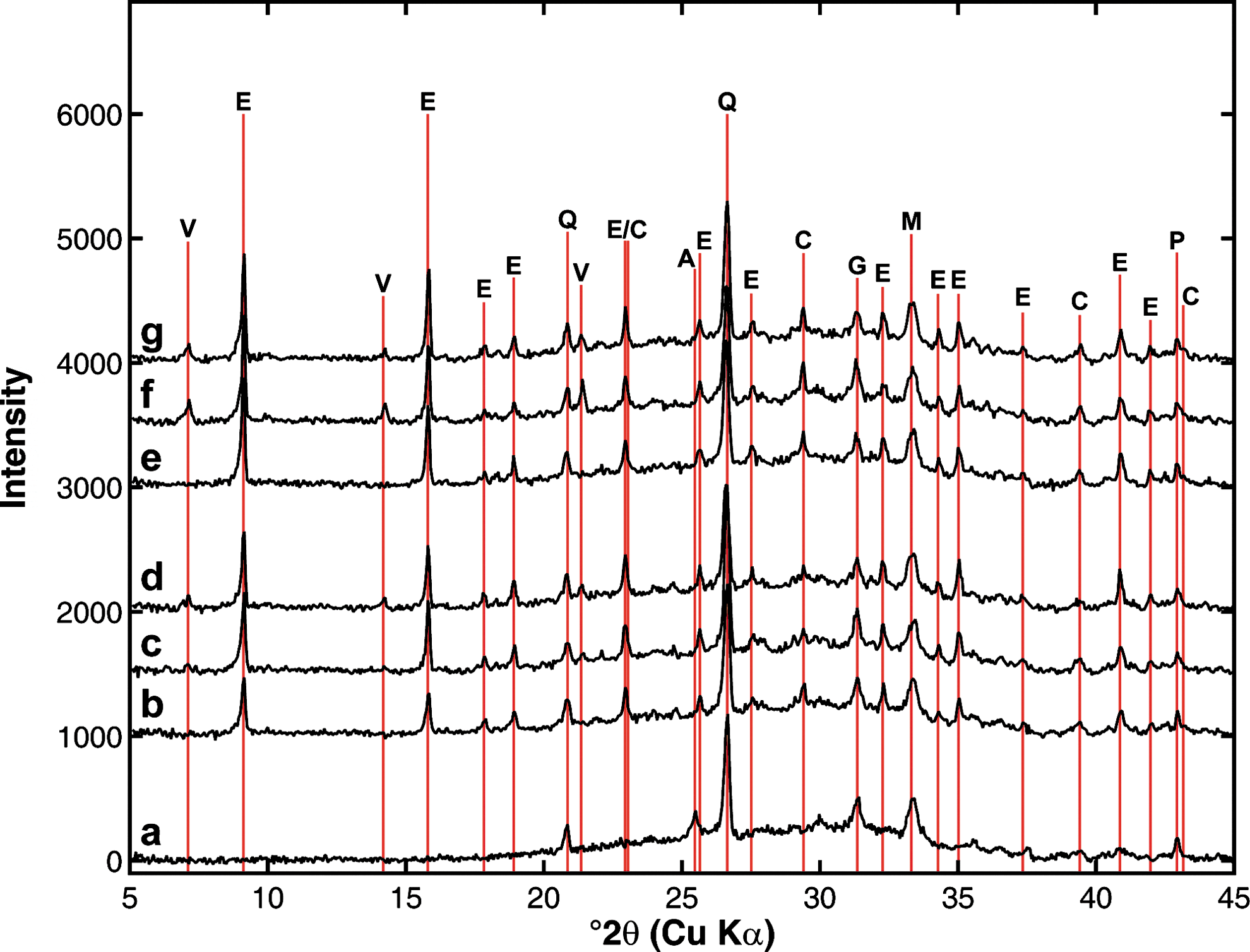
Powder X-ray diffraction (XRD) patterns of
Discussion
The maximum arsenic, selenium, and chromium concentrations measured in the water column of the U.S. ash impoundment experiment exceeded the U.S. primary standards for drinking water (As=10 μg/L, Cr=100 μg/L, Se=50 μg/L), and the selenium and chromium were also at concentrations higher than the Criteria Continuous Concentrations for aquatic life (Cr[VI]=11 μg/L, Se=5 μg/L), which are guidelines for establishment of standards (U.S. EPA, 2012). The concentrations of chromium and selenium reached high values during the initial mixing period, and then slightly increased over the remainder of the experiment. In contrast, the arsenic concentration for the U.S. ash impoundment started at a low value, and then steadily increased over time.
While the total arsenic content of the ash did not change substantially, the solid-phase arsenic speciation may have changed as the amount recovered during the DCB step decreased and the quantity recovered by hot acid digestion increased (Fig. 4). While it is intriguing that the releases of arsenic and aluminum were correlated (Fig. 1), a detailed X-ray absorption spectroscopy investigation of this ash determined that the arsenic was predominantly present as calcium pyroarsenate (Ca2As2O7) or a structurally similar phase (Luo et al., 2011). Because the majority of the arsenic remained in the ash during the ash impoundment experiment, it is possible that the arsenic released to the water column came from the release from a minor Al-rich constituent of the ash that also contained appreciable arsenic. Ultimately, only 0.0015% of the arsenic reached the free water column. The ash has a considerable amount of amorphous phases (Luo et al., 2011) that could include calcium- and aluminum-rich glasses. If arsenic were contained within these phases, then slow dissolution of the phase could have simultaneously released both arsenic and aluminum until they reached stable values after 20 weeks. No correlation of arsenic and aluminum release was seen for the Indian Class C ash.
The release and uptake of calcium observed in the U.S. ash impoundment is related to the dynamics of solid dissolution and secondary solid precipitation. The immediate release of calcium is due to the initial dissolution of lime (CaO) and potentially other Ca-rich solids during the formation of the impoundment. Although the lime readily dissolves, the calcium concentration decreased in the water column during the first 3 weeks of the experiment (Fig. 1) because of reaction with CO2 to form CaCO3. The formation of CaCO3 and the loss of dissolved calcium in the system were indicated by the decreasing pH of the impoundment and the formation of a white precipitate at the surface of the ash layer.
In comparison with the original ash, the total selenium content of the ash declined to roughly half of its initial value and the decrease came almost entirely from the loss of the water soluble fraction of selenium (Fig. 4). The rapid dissolution of lime may have significantly affected the aqueous concentrations of Cr and Se, since the most oxidized forms of these elements (CrO42− and SeO42−) are most soluble at high pH. After the initial release of selenium, the sequential extraction results do not show any further changes in speciation with aging. Interestingly, using the mass balance, only 1.2% of the total selenium can be accounted for by the free water column, which indicates that pore water concentrations in the ash layer are much higher than in the free water column and that diffusion of selenium out of the ash layer can be a significant long-term source.
The chromium concentrations of the ash are essentially the same before and after formation of the ash impoundment, but the overall chromium concentration is much larger compared with selenium, so releases to the water would have had a limited effect on the remaining chromium concentrations. The free water column contained 0.7% of the total chromium initially in the ash. With time, the speciation of chromium in the ash layer may have changed, indicated by an increase in the fraction associated with the heated acid digestion and a decrease in the fraction extractable with DCB. The decrease in extractable chromium could be caused by incorporation into ettringite (Leisinger et al., 2010).
The Indian Class C ash (Indian Ash 1) had high initial water column concentrations of both selenium and chromium. The initial release was likely from the dissolution or desorption of highly mobile species, which may be related to the high initial pH of this impoundment. The concentration of both elements steadily decreased following the formation of the impoundment, which may have been caused by the decreasing pH with time and associated increases in adsorption of selenium and chromium species. Incorporation into ettringite may have also decreased the selenium and chromium concentrations.
The XRD results of the U.S. Class C ash demonstrate that the aging of the ash layer resulted in the formation of minerals (Fig. 6) that may represent new host phases for sequestering or adsorbing trace elements. The redistribution of chromium and arsenic from the more labile fraction that can be extracted with DCB to the residual solids that are only extracted in heated acid may be associated with the formation of these new phases. Despite the dissolution of elements within the ash impoundment and the alteration of the mineralogy with aging, the major element composition of the ash column did not vary during the course of the experiment.
Summary and Conclusions
The evolution of the water column composition of the ash impoundments was strongly dependent on the type of ash studied. The U.S. and Indian Class C ash impoundments had initially high pH and calcium concentrations that declined as the water column took up CO2. At the high pH of the U.S. ash impoundments, selenium and chromium were rapidly released, while arsenic concentrations increased more gradually. The Class F ash impoundments had neutral pH values and lower calcium concentrations. Impoundments with Indian Ashes 2 and 3 had initially high selenium concentrations that declined with time and gradually increasing concentrations of arsenic and chromium. The concentrations of the trace elements exceeded drinking water standards and aquatic life water quality criteria.
Sequential extraction of ash cores from the U.S. Class C ash impoundment provided complementary information to the water column composition. The water soluble fraction of the selenium in the ash was rapidly released, but the remaining selenium speciation did not change. Some correlation between increasing arsenic water column concentrations and declining surface ash arsenic contents were observed. While the overall contents of the trace elements in the ash layer did not change substantially with time, some redistribution of arsenic and chromium from phases that could be extracted with a DCB solution into a more recalcitrant phase that only released the elements in strong acids at 100°C was observed. Changes in the mineralogy of the ash indicate the formation of calcite with time as well as the production of new minerals, which may be related to the increased resistance of the arsenic and chromium to extraction from the ash.
While the results of the study yield insights into the fate of metals in ash impoundments, additional factors should be considered for actual ash management operations. The metal behavior in this study was dominated by chemical processes, but biological growth can influence metal fate and transformations in actual impoundments. Actual impoundments are open systems and their metal concentrations will depend on the rates of metal release from the ash and rates of transfers of ash into and water out of the impoundments.
Footnotes
Acknowledgments
This research was supported by a grant from the Consortium for Clean Utilization at the Washington University. We appreciate the assistance of T.J. Pepping with sampling activities.
Author Disclosure Statement
The authors have no commercial associations that might create a conflict of interest in connection with this manuscript.