
Abstract
Abstract
Complexation kinetics of Co(II), Cu(II), and Ni(II) ions with humic acids was studied. Humic acids were used in three different forms: solid powder, colloidal solution (sol), and hydrogel. Pseudo-second-order rate equation was used as the basic equation for the mathematical description of the complexation kinetics in the humic systems. Three different methods were applied to monitor the concentration changes in studied systems (conductometry, potentiometry, and UV/VIS spectroscopy). A double-linear fit was obtained in all cases. This is an indication that there are two principal types of metal binding sites in humic acids in all used forms. The strongest active centers are probably occupied in the first step, and slower interactions with less preferable binding sites and the rearrangement of formed complex follow in the second step. Values of rate constants were much higher for the first step in all cases. The highest values were obtained for humic sol and Cu(II) ions and the lowest ones for humic solid powder and Ni(II) ions. In general, the complexation rate increased in the following order: humic powder, sol, and hydrogel for all used metal ions and analytical methods.
Introduction
H
One of the conclusions of the 14th Meeting of International Humic Substances Society 2008 is that systematic reactivity mapping studies are needed. Humic substances are chemically reactive, and they possess a wide range of functionalities, which could change when the structural modifications are artificially made to acquire new desired properties. To better understand their chemistry and their possible synthesis/modification/use for environmental remediation, we must understand their exact structure, find appropriate methodologies for the conversion to other materials, and gain fundamental knowledge regarding their chemistry and properties (IHSS, 2008).
One of the main features of humic substances in the environment is their capability of interacting with metal ions to form soluble complexes, colloidal substances, and/or insoluble substances. The contamination of environment by toxic heavy metal ions has become a serious issue because these ions are non-biodegradable and tend to accumulate in living organisms (Zhan et al., 2013). The toxicity of metals and their uptake is significantly decreased by the complexation with humic acid, which has an important role in regulating the metal bioavailability in nature (Soler-Rovira et al., 2010; Wang et al., 2010; Akinci and Ongel, 2011; Kamunde and MacPhail, 2011). Ma et al. (2001) studied the reductive dechlorination of trichloroethene mediated by humic-Ni complexes in a homogeneous aqueous solution. They showed that the humic–metal complexes may be important electron transfer mediators in natural systems. The protective function of the corrosion layer formed on the surface of copper in soil or humic acid electrolytes was observed by Souissi (2013).
Many works deal with the complexation of humic substances using various methods, for example, potentiometric titration (Gamble et al., 1980), voltammetry (Buffle et al., 1987; Carballeira et al., 2000), fluorescence spectroscopy (Senesi, 1990), dialysis (Tipping et al., 1988), X-ray absorption near-edge spectroscopy (Ghabbour et al., 2007), or polarography (Fasfous et al., 2006). Fewer published works studied the complexation of humic substances in the form of hydrogels or aqueous solutions. Martyniuk and Wieckowska (2003) compared the cation exchange capacity of solid and gel forms of humic acids with respect to various metal ions. Metal adsorption on humic gels was greater than that on the solid humic acid samples. The determined metal adsorption capacity of humic gels was three times higher in dynamic conditions compared to the capacity obtained in static conditions, which demonstrated the significant role of their mobility. Ryabova (2008) studied the complexation of Ni(II) and Cu(II) in aqueous solutions with humic acids and their hydroxymethyl derivatives. Town and Powell (1993) stated that the colloidal/particulate humic molecules are stronger complexing agents than the smaller (soluble) humic moieties. The apparent stability of Cu(II)-humic complexes decreased with an increasing metal-to-ligand ratio and with increasing ionic strength. Kislenko and Oliinyk (2003) studied binding of Cu(II), Co(II), and Ni(II) cations to humic acids and their sodium salts in aqueous media. They described the distribution of metal ions between the dissolved phase and the solid phase of humic acids as an equilibrium process depending on the circumstances of performed experiments. They stated that this form of humic acids affects mainly their mobility, but it has no significant effect on the complexation capacity.
This work is focused on the interactions of humic acids with metal ions and their kinetics in various dispersion systems: sol, gel, and suspension (of solid humics). This knowledge is important to evaluate the physicochemical behavior of metal ions in the natural environment. The bioavailability, mobility, and toxicity of metal ions depend on their speciation in natural systems (Garcia-Mina et al., 2004; Bigalke et al., 2010; Kostic et al., 2011). The kinetics of each phase is closely connected with the mobility of molecular entities, which decreases from the sol to the suspended state. Organic matter can enhance (when dissolved) or retard (when in the solid matrix) the transport of contaminants in nature (Benedetti et al., 1995; Weng et al., 2002).
Humic sols can be expected to occur preferably in water systems and to have all or most of their reactive groups or active centers available for the interactions. Humic gels could be found in water sediments, swelled peat, soil, or coal. Some of their functionalities should be engaged in forming the gel structure, but the whole system, including its interior, is still accessible to various interacting particles, penetrating through the gel network. In suspensions, the humic constituents are tightly bonded to the outer surface (including surface of pores) of the solid fraction and essentially only this is accessible for intermolecular interactions. The suspension state can appear in unswollen soil or water streams.
Although the kinetics studies are not ignored within the humic research (e.g., Choppin and Clark, 1991; Anirudhan and Suchithra, 2010; Li et al., 2010; Lin et al., 2011), they are not so numerous as various structural and spectroscopical characterizations or investigations on equilibrium states. Bonifazi et al. (1996) identified the kinetically distinguishable species present in the solution of Cu(II) equilibrated with soil humic acid. The two determined rate constants of complexation consistently represented the middle and slow components of the copper–humic acid complex. The fast component, which reacts on mixing, was the major component present. Rate (2010) stated that the equilibrium modeling approaches to estimating the bioavailability may be insufficient because of the consistency of experimental data with kinetic rather than thermodynamic control.
We believe that the rates of interactions of humic substances and metals, whether they are covalent or noncovalent, are important for evaluating and understanding their role in natural systems and human-driven applications. Despite their heterogeneous mixed character, uncertainties in their structure, and ongoing discussion on polymeric or supramolecular aggregate essence, humic substances are definitely colloids. Thus, besides their polyelectrolyte character, their particular colloidal state may significantly affect their behavior and properties. Indeed, in natural systems, humic substances may occur in different colloidal states.
Experimental
Humic acids were extracted from South Moravia lignite by means of alkaline extraction (Klucakova and Pekar, 2005b). The basic characteristics (based on previous works) are listed in Table 1. More details on the chemical structure of both the original lignite matrix and the isolated humic acids can be found in previously published works (Klucakova and Pekar, 2003a, 2005b, 2006; Peuravuori, 2006).
at. %, atomic percent.
The extracted humic sample was finely milled and used directly for complexation in the form of humic powder or for the preparation of humic sol and hydrogel.
Humic sols were prepared by the dissolution of the humic acid powder in the 0.1, 0.5, and 1 M NaOH solutions. To neutralize NaOH, the appropriate amount of HCl of the same concentration as NaOH was added to each solution. The resulting sols had the concentration of humic acids of 0.5 g/L.
The humic gel was prepared from the solution of humic acids in 0.5 M NaOH (8 g/L) by precipitation with the concentrated HCl solution (∼35% wt.). The gel was washed with deionized water and centrifuged repeatedly to remove Cl− ions. The content of humic acids in the gel was 12% (wt.).
These three prepared forms of humic acids (the solid powder, the colloidal solution [sol], and the hydrogel) were further used for the studies of the complexation of humic acids with Co(II), Cu(II), and Ni(II) ions. Co(II), Cu(II), and Ni(II) ions were used in the form of 0.05 M solutions of their anhydrous chloride salts (99.99% trace metal basis). The individual humic forms for the complexation experiments were prepared in the following manner: 1 g of humic powder was mixed with 50 mL of 0.05 M Me2+ solution; the humic sol was mixed with 0.05 M Me2+ solution in the ratio of 25 mL/25 mL; 1 g of hydrogel was mixed with 50 mL of 0.05 M Me2+ solution. The systems were stirred and the conductivity, the pH, and alternatively, the absorbance were monitored, as described in the following text.
The complexation kinetics was studied using several methods. A Sentron Titan K185-016 pH meter with an ISFET sensor was used for the monitoring of the production of H+ ions during the complexation. The range of recorded values was between 2 and 8. A Hanna Instruments HI8820N conductometer with the HI 7687 platinum 4-ring conductivity probe and 0–100 mS/cm conductivity range was used for the measurement of conductivity. An UV/VIS Hitachi U3900H spectrometer was used for the determination of the decrease in concentration of free metal ions in studied systems. Absorbance was measured in quartz cuvettes (10 mm across) at the wavelengths of 510 nm (Co), 720 nm (Ni), and 810 nm (Cu). The concentrations of free metal ions in studied systems were calculated on the basis of the decrease in absorbance and calibration curves. The UV/VIS spectroscopy was used for the monitoring of kinetics only in the case of humic powder.
All experiments were done in triplicate, and the results presented in this work are the average values.
Results and Discussion
To compare the kinetics in various humic dispersions, the widely utilized pseudo-second-order rate equation (Moore, 1999; Atkins and de Paula, 2009) was used as the basic equation for the mathematical description of the complexation kinetics in all treatments:
where k is the rate constant of the second-order reaction, x0 is the initial concentration of humic binding sites, y0 is the initial concentration of metal ions, and z is the concentration of formed metal–humic complexes. The pseudo-second-order rate equation corresponds with the summary reaction of one metal ion with one binding site of humic acids.
The integrated form of Equation (1) is
Equation (2) was directly used for data processing in the case of complexation in the humic suspended state and the spectrophotometrical measurement. The concentration of formed metal–humic complexes (z) was determined as the decrease in concentration of free metal ions in the solution. The rate constant is denoted as kA in this case.
For conductometric and potentiometric data obtained from the measurement of all humic dispersion forms, Equation (2) was modified to the following forms (Klucakova and Pekar, 2003b, 2004, 2005a).
In the case of second-order reaction with y0>x0, the binding sites in humic acids become gradually exhausted, therefore, their concentration in time t→∞ is equal to zero, the concentration of free metal ions is equal to y0-x0, the concentration of metal–humic complexes is equal to x0, and the concentration of formed H+ ions is equal to 2x0. In view of the fact that the mobility of H+ ions is much higher than the mobility of free metal ions, the increase in conductivity during the complexation is caused mainly by splitting off the H+ ions from humic binding sites (other influences on the conductivity are neglected). On the basis of these assumptions, we can write the following relations:
and
The combination of Equations (2)–(4) gives the resulting form for the conductometric data:
where G0, Gt, and G∞ are the initial conductivity, the conductivity in time t and in time t→∞ , respectively, and const can be expressed as follows:
According to the above-mentioned assumptions and proposed summary reaction of one metal ion with one binding site of humic acids, we can write that the amount of formed metal–humic complexes is directly proportional to the increase in concentration of H+ ions from humic binding sites, that is, z=ht−h0. Therefore, Equation (2) can be modified for potentiometric data as follows:
where Δh is the increase in concentration of H+ ions in given time (Δh=ht − h0). The rate constant k of the second-order reaction was marked in modified Equations (5) and (7) as kB and kC. For more details regarding the derivation of Equations (5) and (7), please see our previous works (Klucakova and Pekar, 2003b, 2004, 2005a).
The extrapolation of values obtained for humic sols was carried out to compare them with other humic systems (so-called primary salt effect):
where kI is the rate constant for sol with the ionic strength I, za and zb are the charges of reacting ions (Moore, 1999; Atkins and de Paula, 2009).
As expected, the mechanism of complexation of humic acids with used metal ions is more complex than a simple second-order reaction. Two main steps were observed in measured kinetic data for all treatments (Fig. 1). We have found that two main steps can be observed for all methods used for the monitoring of complexation kinetics. The values for humic sol are obtained by the extrapolation on the ionic strength I=0 according to Equation (5), because humic sols are prepared on the basis of humic acids in the NaOH solution neutralized by HCl. The example of extrapolation for the first complexation step is shown in Fig. 2. The results in Tables 2–4 can be characterized by several basic features. The kinetic constants for the first complexation step are much higher. The values determined for Cu2+ ions are the highest in most cases. The constants decrease in the following order: humic sol, humic hydrogel, humic powder. While the rate constants kB and kC for the first complexation step are comparable in the case of humic sol and humic gel, kB values are lower than kC ones for humic powder. The constants kA are much higher than the values determined by other used methods.
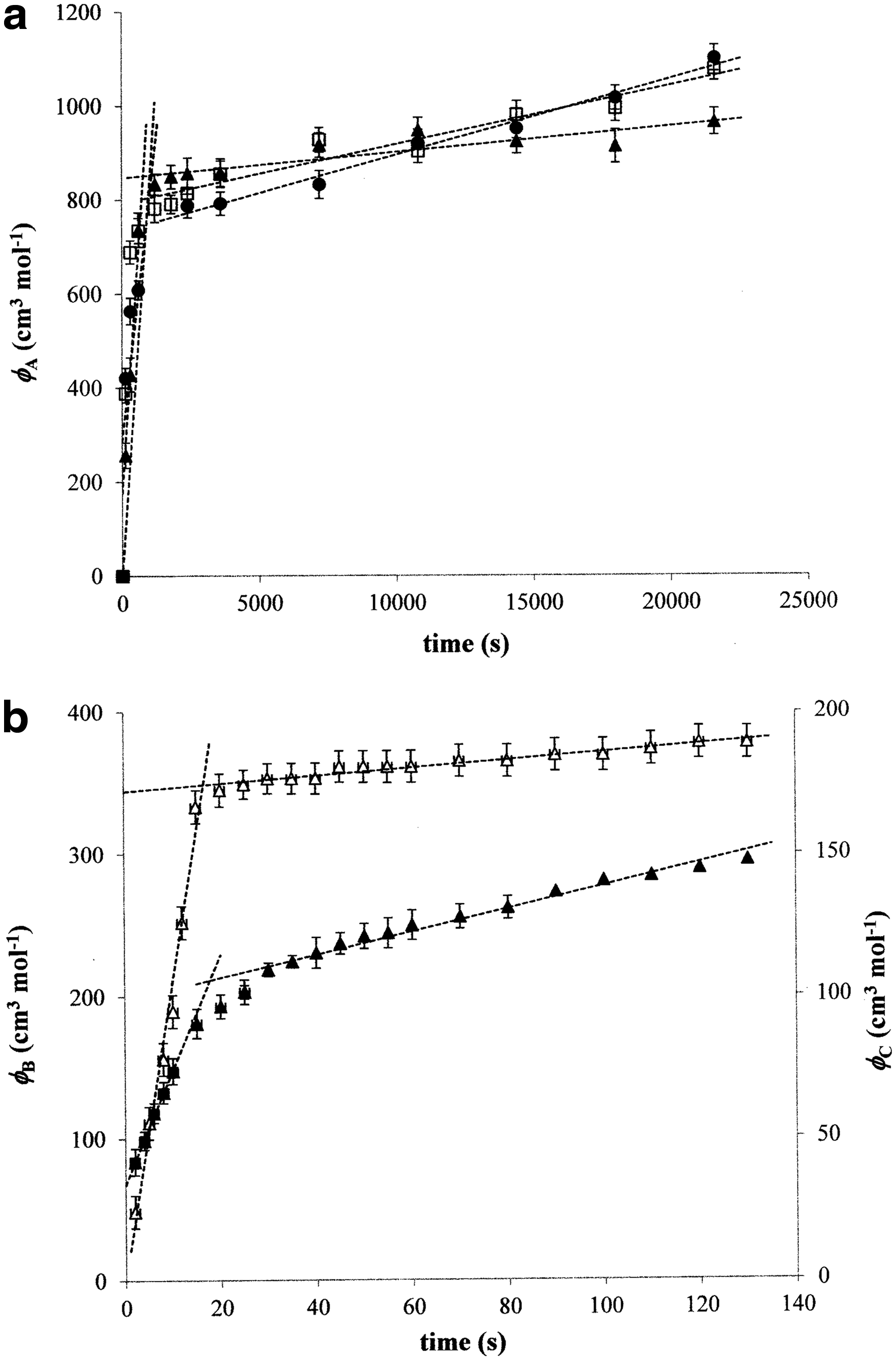
Examples of experimental data fitted by the kinetic equation of second-order reaction:
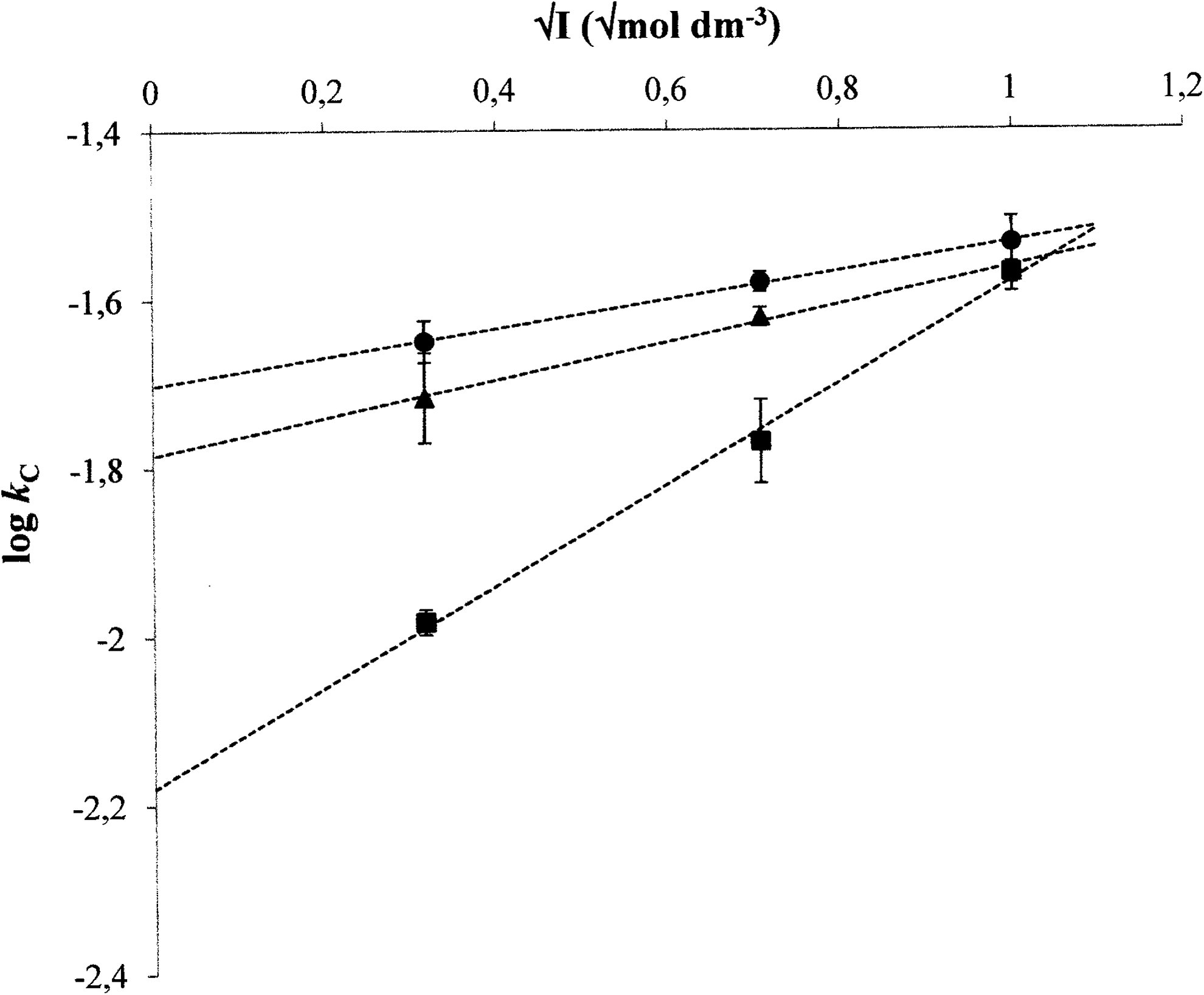
Experimental data obtained for the interactions of Co2+(triangles), Cu2+ (circles), and Ni2+ (squares) ions with the humic sols with various ionic strengths in the first complexation step fitted by Equation (8).
Values of k′A were determined using Equation (2) and differences between data obtained by spectrophotometry and potentiometry.
nd, not determined.
The double-linear fit was obtained for all experimental data. This is an indication that there are two principal ways for the metal complexation with humic acids in all used dispersion forms. The intersections of the linear sections range from minutes for humic powders or gels to seconds for humic sols. This intersection probably means the point at which the complexation mechanism shifts. A relatively high fraction of metal ions (60–80%) are bonded with humic acids in these intersections. This could signify that the majority of metal ions are complexed relatively quickly (independently of the type of humic dispersion) and their residual part is then complexed at a lower rate. The binding sites, which are the most attractive for metal ions, are occupied probably in the first step (Jin et al., 1996; Ma et al., 1999). They become gradually exhausted and the active centers with lower affinity are occupied in the second complexation step. According to some published results (Jin et al., 1996; Ma et al., 1999; Klucakova and Pekar, 2006), the binding sites of humic acids could be divided into two groups: strong and weak and the intersection could be related to the exhaustion of strong sites and to the start of interactions of weak ones. The intercept is then a hypothetical value of ΦA and ΦC for this change in complexation mechanism. In the case of ΦB, the intercept included also the values of initial conductivity (G0) and conductivity after the exhaustion of humic binding sites (G∞).
The individual methods are differently sensitive to different processes in the complexation, as described below. The spectrophotometry measures the decrease in concentration of metal ions in the source solutions of metal ions. The amount bonded to humic acids determined by this method is the total amount complexed by humic active centers of various types. The amount measured by potentiometry represents the metal ions bonded to active sites capable of splitting off hydrogen ions during the complexation (e.g., carboxylic and phenolic groups). The data obtained using the conductometry connected two opposite influences: the decrease in conductivity caused by the decrease in concentration of free metal ions in the solution and the increase in conductivity connected with the liberation of hydrogen ions as the result of the complexation of metal ions with acidic functional groups of humic acids. Hence, the conductometry is sensitive to both, the total decrease in concentration of free metal ions and the production of hydrogen ions as a consequence of the dissociation of some functional groups during the complexation.
It is not possible to use UV/VIS spectrophotometry for monitoring of complexation of humic sols and gels because of the strong brown color of measured solutions. Therefore, we can compare all three humic dispersions using only the conductometric and potentiometric data. The rate constants obtained on the basis of these two methods differ for individual dispersion systems. The highest values were obtained for humic sols, which correspond with the highest complexation rate and better accessibility of binding sites in dissolved humic form.
The kinetic experiments in humic sols revealed another interesting fact. We assume that the increase in conductivity is caused mainly by the production of hydrogen ions from humic acidic groups during the complexation, because the mobility of these ions is much higher in comparison with other ions in the system. Therefore, the time development of conductivity should correspond with that of pH. However, it was found that the conductivity increased also after the pH was stabilized. This phenomenon was observed in the case of complexation of Cu2+ and, partially, Co2+ ions. The explanation could be that metal ions occupied the maximum of active centers in the beginning stage and then a rearrangement in the structure of the formed complex followed. Because bivalent metal ions need two functional groups, it is necessary to find them not far from each other to form the stable complexes, which can result in the liberation of a part of metal ions causing the conductivity increase. The phenomenon for Cu2+ ions was observed and described in detail in our previous works (Klucakova and Pekar, 2003a, 2005a). The comparison of the kinetic rate constants for humic sol in the first and second complexation steps (Table 2) showed a relatively strong decrease in values determined on the basis of potentiometric data. It means that the production of hydrogen ions decreased in this step and the conductivity increase is probably caused rather by the liberation of metal ions during the rearrangement. It is also the reason for higher kB values in comparison with kC ones. Although the changes of conductivity and pH stop at the same time in the case of humic gel, the reason for noticeable differences between the values of kB and kC in the second complexation step could be similar.
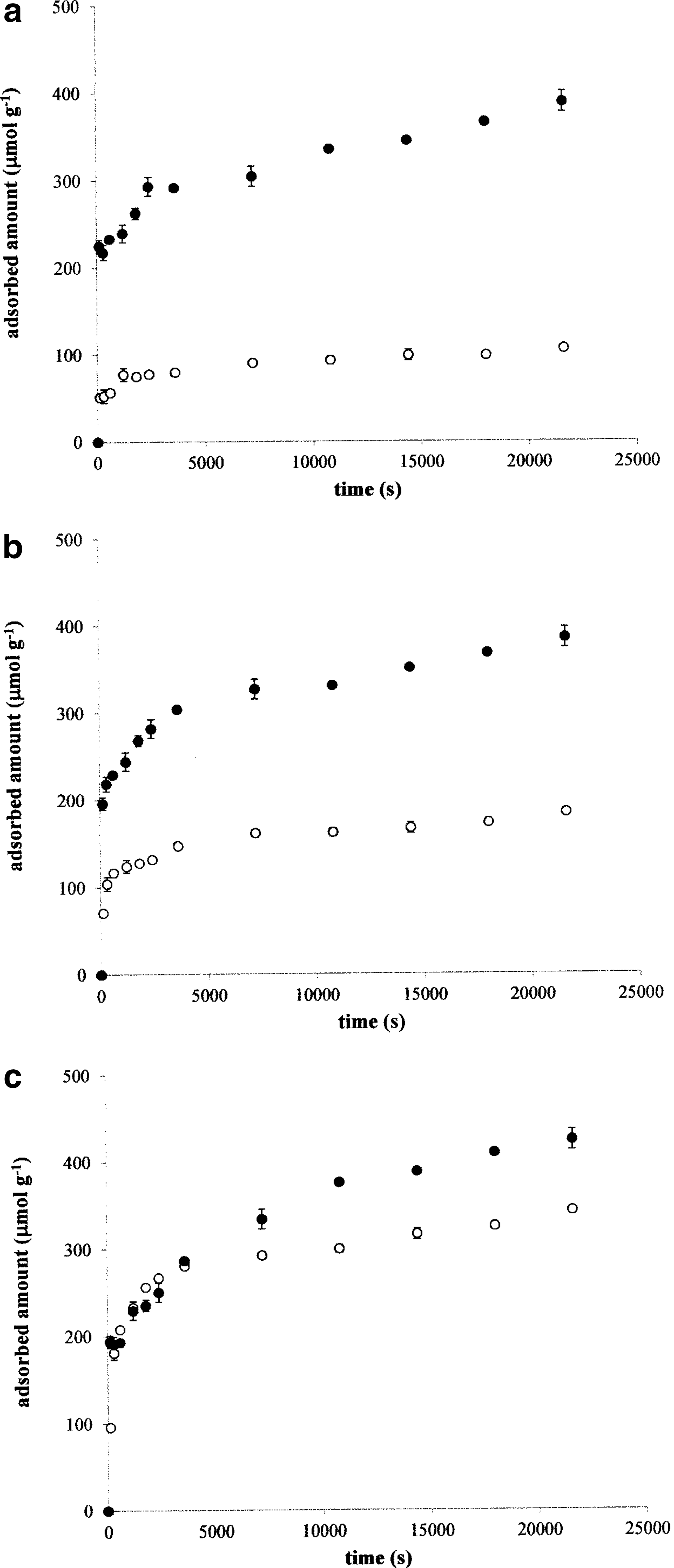
To study the complexation of humic acids with metal ions in detail, we used the UV/VIS spectrophotometry for the determination of total decrease in the concentration of metal ions in the sorption experiments (Figs. 3 and 4). Comparing the kinetic data obtained from spectrophotometry and potentiometry, it can be found that a part of metal ions bound with humic acids is bonded to humic active centers without the production of hydrogen ions. It is well known that metal ions can be bonded to humic acids through various active centers. The results of our previous work (Klucakova and Pekar, 2006) showed that two basic types of humic binding sites can be defined. The first type is characterized by the ability to split off hydrogen ions in complexation and includes acidic functional groups (mainly carboxylic and phenolic). Other binding sites such as phenolic ethers (Baker and Khalili, 2003), aromatic structures, O-alkyl groups in side chains (Fuentes et al., 2013), or acidic functional groups dissociated before the complexation do not produce hydrogen ions. Listhvan et al. (2009) stated that copper can be bonded to humic acids without the participation of functional groups. Although the authors did not study other metals, our results indicate that this macrocoordination could be possible also for them.
Kinetic data obtained for sorption of Cu2+ ions on humic powder using potentiometry (white) and UV/VIS spectrophotometry (black) at 25°C
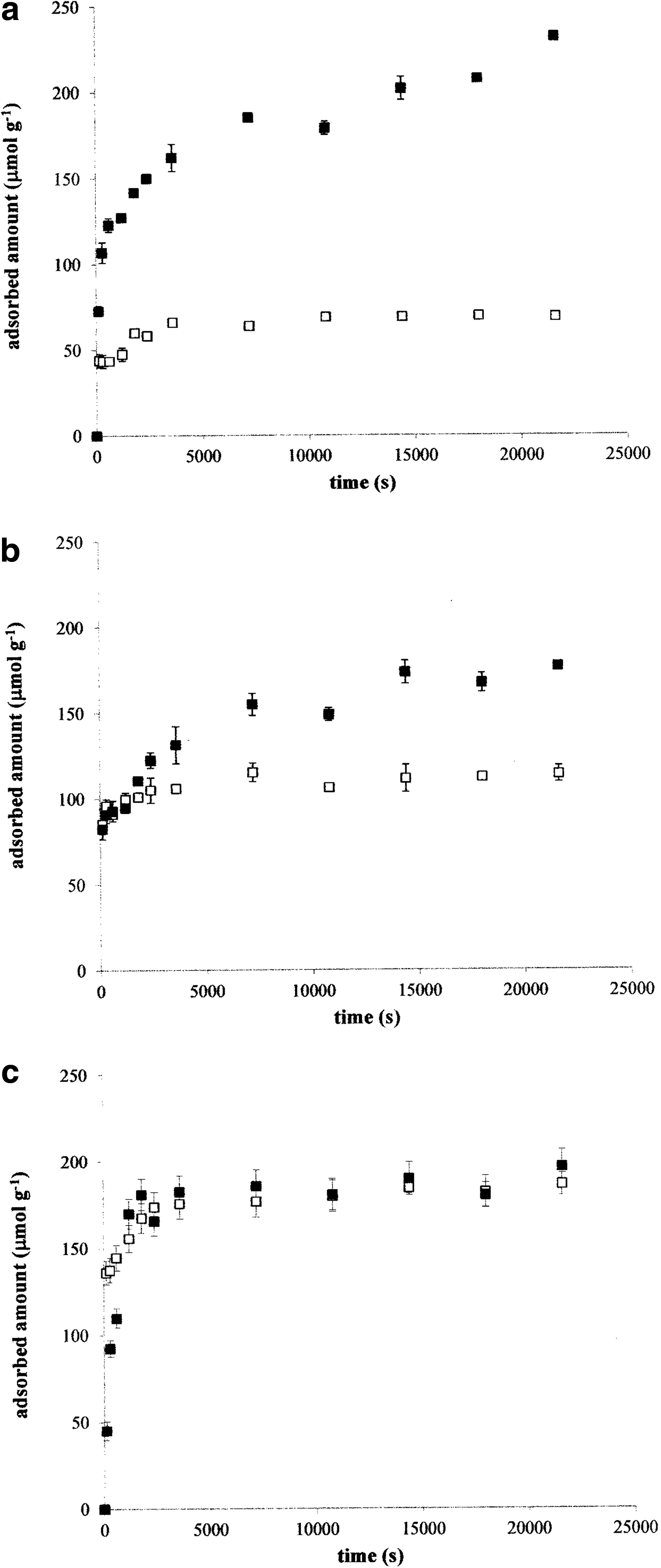
Kinetic data obtained for the sorption of Ni2+ ions on the humic powder using potentiometry (white) and UV/VIS spectrophotometry (black) at 25°C
To determine the rate constants for the binding sites that are not able to liberate hydrogen ions, the differences between spectrophotometric and potentiometric data were used (adsorbed amounts determined by UV/VIS spectroscopy were subtracted from the amounts determined by potentiometry). The rate constants k′A were calculated using Equation (2) (Table 4). The sorption experiments at higher temperatures showed their different influences on the kinetic data obtained on the basis of various methods. The data for Cu2+ and Ni2+ ions in Figs. 3 and 4 confirmed also the different influences on individual metal ions. We can see that the amount of produced H+ ions increased with the temperature relatively strongly in the case of both metal ions and a similar progress was observed also for Co2+ ions. The increased production of hydrogen ions resulted in the increase in measured conductivity, which corresponds with much higher mobility of these ions in comparison with other ones.
The time development of the total decrease in concentration of metal ions in the solutions surrounding the humic powder and the total amount of formed metal–humic complexes differed with the type of metal ion. In the case of Co2+ and Cu2+ ions, the complexation rate and the amount of formed complexes increased (but only imperceptibly). In the case of Ni2+ ions, the complexation rate as well as adsorbed amounts measured spectrophotometrically decreased gradually with increasing temperature. The data obtained on the basis of UV/VIS spectrophotometry and potentiometry are practically the same at 70°C (Fig. 4c); therefore, the values of k′A could not be calculated (the differences between spectrophotometric and potentiometric data were equal to zero). In all other cases, the differences between spectrophotometric and potentiometric data decreased with increasing temperature, which resulted in the decrease in the rate constants. While the increasing temperature supported the complexation with acidic functional groups, it suppressed the adsorption on other binding sites.
Summary
The kinetics of complexation of metal ions with humic acids in three various dispersion forms—solid powder, colloidal solution (sol), and hydrogel—was studied using conductometry, potentiometry, and UV/VIS spectrophotometry. Two main steps were observed in the measured kinetic data for all treatments and for all methods used for the monitoring of complexation kinetics. Our results showed that the majority of metal ions are complexed relatively quickly and their residual part is then complexed at a lower rate. The binding sites of humic acids could be divided into two groups: strong and weak and the change in complexation mechanism represented by the intersection in Fig. 1 could be related to the exhaustion of strong sites and the interactions with weak ones. Experimental data were fitted by the kinetic equation of second-order reaction. The rate constants decreased in the following order: humic sol, humic gel, humic powder. In the case of humic powder, it was found that metal ions can be adsorbed not only by acidic functional groups but also by binding sites that are not able to split off hydrogen ions. The adsorption on these binding sites is suppressed by increasing temperature in contrast to the complexation by acidic functional groups.
Footnotes
Acknowledgment
This work was supported by the Ministry of Education, Youth, and Sports: Project No. LO1211.
Author Disclosure Statement
The author declares that no competing financial interests exist.