
Abstract
Abstract
Since nanoparticles are considered emerging contaminants in water, there is a pressing need to ensure their removal. Specifically, recent studies have found titanium dioxide nanoparticles (TiO2) to potentially cause adverse environmental and health effects. Typical treatment plants have three stages of prefiltration treatment in which particles may be removed: coagulation, flocculation, and sedimentation. Scaled-down jar tests, with constant dosage of aluminum sulfate as the coagulant, were utilized to investigate the efficacy of conventional treatment facility to remove nanoparticles and the fundamental mechanisms involved under ideal and environmentally relevant conditions. Results showed that removal by sedimentation was the most efficient in the presence of divalent cations and in the absence of natural organic matter (NOM) and synthetic organic coating on the nanoparticles, achieving >1 log removal. With the addition of a synthetic organic coating on the nanoparticles and NOM, removal decreased to ∼80%. Despite the high dosage of coagulant used, without pH modification (typical of practical treatment), under realistic water chemistries there is a possibility for significant release, and specifically ∼5 ppm of particles smaller than 450 nm were observed after sedimentation, which raises concern regarding the ability for these engineered treatment facilities to adequately remove these nanoparticles that are increasing in production and use.
Introduction
E
There is significant concern of potential health and environmental risks associated with these emerging nanomaterials such as TiO2. Some studies have found minimal toxicity related to bare TiO2 nanoparticles, while other studies have found adverse health effects such as oxidative stress in human cells (Gurr et al., 2005; Long et al., 2006; Warheit et al., 2006; Vicario-Parés et al., 2014). It is well documented that TiO2 (in nanoparticle or colloidal form) is toxic to various organisms within surface water environments due to the formation of reactive oxygen species, in the presence of UV light, that will nonselectively oxidize organic compounds (Bar-Ilan et al., 2012, 2013; Dalai et al., 2012). However, most ENMs have some form of an organic coating either as the active material or are used to suspend or stabilize the materials (Shen et al., 2005; Aitken et al., 2006; Chen and Mao, 2007; Brar et al., 2010; Macwan et al., 2011). There is a significant increase in toxicity of TiO2 nanoparticles with such organic coatings (Vicario-Parés et al., 2014). Removal of such ENMs via water treatment facilities under environmentally relevant conditions is of particular interest, as effluent is either discharged directly to surface water or distributed through our drinking water supplies.
Numerous studies investigate the transport of TiO2 nanoparticles and other ENMs within the environmentally relevant conditions such as in the presence of natural organic matter (NOM) and divalent ions. The presence of NOM has been observed in many studies to affect the behavior of ENMs. Predominately in simple systems (KCl electrolyte), the NOM will increase the stability due to electrostatic and electrosteric repulsion (Thio et al., 2011; Chowdhury et al., 2012), (Keller et al., 2010; Stankus et al., 2010); however, in the presence of divalent ions, the stability is reduced due to calcium bridging (Chowdhury et al., 2012). NOM, at low concentrations, has also been observed to form stable nanoscale colloidal aggregates of extremely hydrophobic materials that were otherwise thought not to be a contaminant in aquatic environments, such as carbon fullerenes (C60) (Fortner et al., 2005; Chen and Elimelech, 2007), multiwalled carbon nanotubes (Hyung and Kim, 2008), or TiO2 with hydrophobic organic coatings (Shen et al., 2005; Aitken et al., 2006; Chen and Mao, 2007; Brar et al., 2010; Macwan et al., 2011). Based on these studies, it has been demonstrated that NOM plays a vital role in the behavior of nanomaterials by reducing the size of aggregates, contributing to steric and electrostatic effects, and increasing particle stability (Petosa et al., 2010).
In addition to NOM, another factor that influences particle stability and aggregation as well as toxicity is the synthetic organic coating for commercial and industrial applications. The Chen et al. study demonstrated that hematite nanoparticles coated with alginate had enhanced aggregation rate in the presence of divalent cations as compared with the monovalent. This was attributed to the divalent cations bridging the alginate and leading to larger alginate-hematite clusters. (Chen et al., 2006, 2007). In contrast, fullerene (C60) nanoparticle deposition rates (determined by Quartz-Crystal Microbalance [QCM-D]) were significantly reduced in the presence of both humic acid and alginate-coated silica surfaces due to steric repulsion as compared with bare silica surfaces (Chen and Elimelech, 2008). In industrial applications, nanoparticles are stabilized against aggregation through the adsorption of coatings (Joo et al., 2009). A coating of particular interest is meso-2,3-dimercaptosuccinic acid (DMSA), which has been used in a variety of biomedical and environmental applications, such as a treatment for lead poisoning (Aposhian and Aposhian, 1990; Asiedu et al., 1995). DMSA acts a chelating agent that is able to bind metal ions to form a complex ring structure called chelates, which possess ligand binding atoms that can form one or two covalent bonds (Crittenden and Harza, 2005). DMSA forms strong complexes with the surface layer of the iron-oxide (maghemite) nanoparticles and increased particle stability in intracellular uptake due to unbound carboxylate groups (Wilhelm et al., 2003). Other studies have demonstrated the use of DMSA as a capping agent for quantum dots (Sevinc et al., 2012) and iron-oxide nanoparticles (Kima et al., 2007; Chen et al., 2008). These studies show the importance of investigating nanoparticle fate in the presence of a coating; however, there are limited studies investigating the impact of synthetic organic coatings on the particle stability in an aquatic setting—specifically in the presence of NOM. Studies under these environmentally relevant conditions with a model coating and humic acid are critical to properly assess the fate and removal of TiO2 that may be present in treated effluent.
Most studies are concerned with the transport of such EMNs within the subsurface or deposition studies, however few, focus on the ability of convention treatment processes to remove ENMs (Fortner et al., 2005; Chen and Elimelech, 2007; Chowdhury et al., 2012). Since many of these ENMs are being used by consumers and therefore being disposed indirectly or directly into our wastewater treatment facilities and contaminating potential drinking water sources, conventional treatment processes are our last line of defense before ingesting such materials (Aitken et al., 2006; Nowack and Bucheli, 2007; Brar et al., 2010; Keller et al., 2013). Conventional water treatment methods, which exploit aggregation-related phenomena (i.e., coagulation, flocculation, and sedimentation) are known to remove NOM, suspended solids, disinfection by-product precursors, and other inorganic constituents from water and wastewater (Duan et al., 2002; Domínguez et al., 2005; Beltrán-Heredia et al., 2009; Zhao et al., 2009; Kim et al., 2012). It has been reported that metal salts (i.e., aluminum, iron) are effective in removing colloidal particles and dissolved organic substances through charge neutralization and sweep flocculation mechanisms, however, are affected by water chemistry and the presence of NOM (Duan and Gregory, 1996, 1998, 2003; Gregory, 2006). One study investigated the removal of C60 stabilized by NOM in which coagulant dosages for sweep flocculation were required under high alkalinity levels for removal (Hyung and Kim, 2009). Such high alkalinity levels are not representative of natural waters that are entering water treatment facilities. A large percentage of the publications are focused on the production of these and new ENMs, and some are focused on their potential toxicity; however, very few are focused on conventional water treatment processes' removal of such materials (Brar et al., 2010). Therefore, additional studies need to be performed under environmentally relevant conditions to truly evaluate the ability of conventional methods to remove such nanoparticles in the presence of NOM as well as with synthetic organic coatings relevant to the applications.
Therefore, the aim of this work was to evaluate conventional water treatment processes, coagulation, flocculation, and sediment, ability to remove a model, and most prevalent ENM TiO2. A previously evaluated scaled-down jar test protocol (Honda et al., 2014) was used to assess the removal using a common coagulant, alum, (Crittenden and Harza, 2005). The objectives were to investigate the impact of ideal (KCl systems) and simulated (artificial ground and surface water) source waters, synthetic organic coatings (modeled using DMSA), and the presence of NOM on the removal of TiO2 at each treatment stage, and to determine the possible mechanisms involved. These relevant aquatic and nanoparticle parameters were considered in this study to gain knowledge of the level of removal by current technologies.
Materials and Methods
TiO2 nanoparticle preparation
Uncoated (bare) titanium dioxide (TiO2) nanoparticles employed in this study were P25 Evonik Degussa (Evonik Industries AG). This TiO2 is an industrial grade nanoparticle that has a phase composition of 18% rutile and 82% anatase with a purity of 99.5%. The average size reported by the manufacturer to be 21 nm (which represents the average size of the anatase and rutile crystals typically measured via X-ray diffraction), however, is composed of fused aggregate particles that can aggregate to form larger secondary aggregates in suspension. Before jar test experiments, a stock suspension of TiO2 was prepared via a similar protocol by Chowdhury et al. (2011). The TiO2 nanoparticles were sonicated (Transsonic 460/H; Barnstead Lab-Line) for ∼2 min to help break up aggregation immediately before jar test experiments. Coated TiO2 nanoparticles were prepared by a similarly reported procedure (Maurizi et al., 2009) by utilizing DMSA as the coating agent for P25 TiO2.
Test solutions
Four solution conditions were tested in this study: two ionic strengths (IS) of KCl, artificial groundwater (AGW), and artificial surface water (ASW). Monovalent electrolyte solutions (KCl) were used in this study at two different IS, 1.83 and 10 mM. These IS values were selected to compare with the IS of ASW (Yip et al., 2011) and AGW (Bolster et al., 1999), respectively. The pH for all solutions was adjusted to 8±0.1 using 0.1 M KOH to mimic the pH of the AGW and ASW recipes used in this study. These simple monovalent electrolyte test solutions were created as a baseline comparison for the more complex source water recipes utilized. The complete list of constituents for AGW and ASW can be found in Table 1. All chemicals were either ACS grade reagents (purchased from Fisher Scientific) or research grade (from Mallinckrodt Chemical and Acros Organics). For select experiments, 1 mg/L of dried Suwannee River humic acid (SRHA) (International Humic Substances Society) was used as the model NOM. Other studies have also used SRHA as a representative NOM (Stankus et al., 2010; Chowdhury et al., 2012) in TiO2 and gold nanoparticle stability tests. This concentration was selected, as it represents a typical average of NOM present in groundwater and surface water (Crittenden and Harza, 2005).
Ionic strength (IS) of the final solutions is displayed in the table.
Jar test experiments
Scaled-down jar tests (Honda et al., 2014) were used to simulate conventional coagulation, flocculation, and sedimentation processes. Aluminum sulfate (or alum, Al2(SO4)3) at a dosage of 50 mg/L Al2(SO4)3•18H2O (75 μM Al2(SO4)3) was used with the intent of sweep flocculation, which is more commonly used for practical water treatment operations (Gregory, 2006). The nanoparticle concentration (bare and coated) was 100 mg/L for all experiments. 1.5 mL samples were taken from the center point of the beaker at ∼1 cm in depth in the beakers at the end of each stage of treatment for jar test experiments. Samples collected were used to evaluate particle concentration, zeta potential, and size. The samples taken at times 0, 30, and 90 min correspond to the end of the three stages of treatment, where the absorbance was measured: 1 min for flash mixing, 30 min of flocculation, and 1 h of sedimentation. Samples were measured in a UV-Vis spectrophotometer (DU Beckman Coulter) at a wavelength of 370 nm. Other studies have successfully employed the use of a UV-Vis to measure the relative concentrations of nanoparticles (Keller et al., 2010; Dalai et al., 2012). Each experiment was performed in triplicate.
Nanoparticle characterization
Electokinetic characterization (zeta potential) and particle sizing (hydrodynamic diameter) was conducted using a ZetaPals Analyzer (Brookhaven Instruments Corp.). Zeta potential was determined from the Smoluchowski equation, which is applicable when the Debeye length (thickness of the double layer) of a particle is much less than the particle size (Elimelech et al., 1998; Gregory, 2006). Measurements were conducted immediately after the completion of each of the three treatment stages. Particle sizing was measured using dynamic light scattering (DLS; Brookhaven Model-BI-9000AT). The zeta potential and hydrodynamic diameters were determined from the average of 5 runs, with each run lasting for 2 min. Two milliliters of test samples were needed for characterization experiments. In total, 3.5 mL of samples are needed for measurements: 1.5 mL for absorption+2 mL for characterization. Inductively coupled plasma mass spectrometry (ICP-MS) was used to determine the residual concentration of TiO2, which is smaller than 0.45 μm. The samples were filtered using 0.45 μm filter and were then acidified using concentration nitric acid. The samples were left for 24 h before analysis using an Agilent 7700 series ICP-MS.
Results and Discussion
Effects of IS and solution chemistry on removal of bare TiO2 nanoparticles
Removal of the bare TiO2 was more effective in the complex waters (AGW and ASW, 97.6%±1.7% and 98.7%±1.5%, respectively) as compared with the monovalent systems (1.83 and 10 mM KCl, 77.0%±5.9% and 85.9%±7.2%, respectively) in the presence of 50 mg/L alum (75 μM Al2(SO4)3). After the addition of the coagulant, the system becomes more acidic (pH ∼7) due to the formation of sulfuric acid from the hydrolysis of alum to form hydrolyzed cationic species (Honda et al., 2014). The dominate species is Al(OH)4−, although a minor species of Al(OH)4+ is also present. The positively charged hydrolyzed species will strongly adsorb to the negatively charged TiO2 particles (IEP ∼ pH6.2) (Duan and Gregory, 1998), leading to charge neutralization and particle destabilization. With a further increase in alum loading, the particles can develop an excess charge from the positive species and this can lead to charge reversal, restabilizing the particles (Duan and Gregory, 1998, 2003). It has been found that only ∼5 μM Al/m2 particle surface is required for charge neutralization at pH6 (Gregory, 2006). Assuming a complete available titanium surface area, there is a minimum loading 55 μM Al/m2 particle surface (∼10% of which is a hydrolyzed cationic species). Therefore, the observed positively charged particles (Fig. 1B) are due to charge reversal from excess absorption of the positively charged Al species.
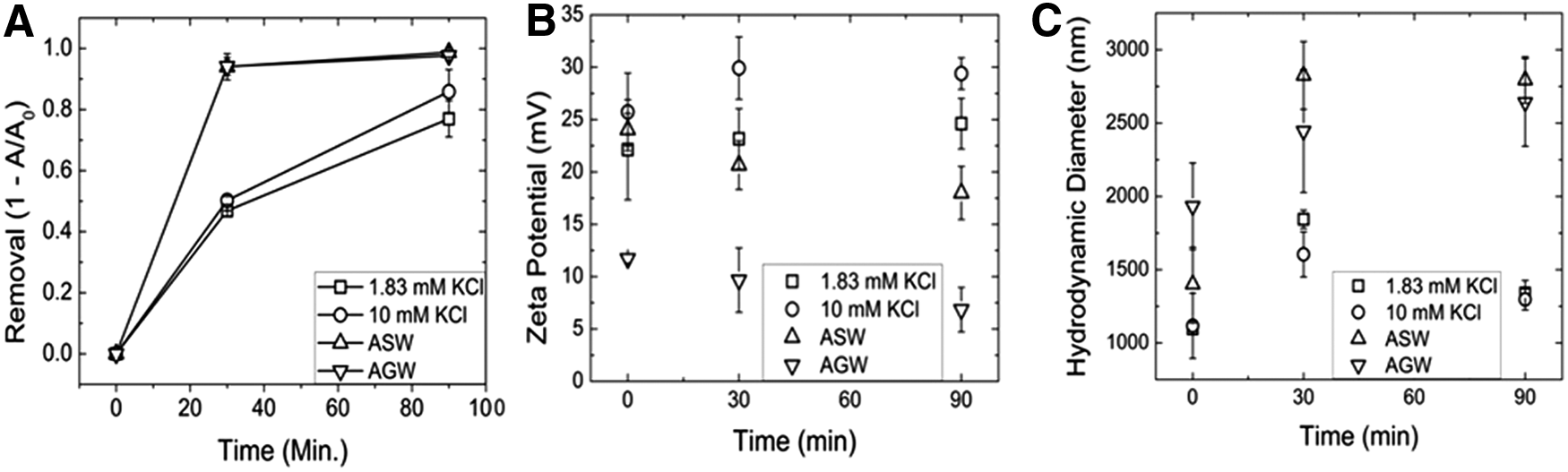
Bare TiO2 removal was plotted in
In addition, it is likely that the excessive coagulant dosage (75 μM Al2(SO4)3) in the concentration of Al (III) exceeds solubility limit (minimum at near neutral pH) and will promote precipitation of amorphous colloidal hydroxide particles (homogeneous or heterogeneous) (Duan and Gregory, 2003). These hydroxide particles have a PZC around pH8 and therefore should be positively charged at near neutral pH (Duan and Gregory, 2003). A combination of these cationic species and positively charged hydroxide particles results in rapid charge reversal of the titania particles and the observed positive zeta potential of the particles (Fig. 1B). With sufficient hydroxide precipitation particles can become enmeshed within the hydroxide precipitate, which is known as “sweep” flocculation. Practical water treatment typically uses such high dosages to ensure extensive precipitation, resulting in high particulate removal (Elimelech et al., 1998; Duan and Gregory, 2003). Sweep flocculation typically occurs above 40 μM Al2(SO4)3.
Increasing IS (within each respective system, KCl vs. simulated waters) did not result in a significant increase in removal efficiency (p>0.05). There was a significant increase in removal between the simple KCl systems and the simulated (AGW and ASW) systems (p<0.05). With regard to the water chemistries, the simple systems (KCl) resulted in significantly more stable suspensions, 24.6±2.4 and 29.4±1.5 mV for 1.83 and 10 mM KCl, respectively, than the more complex waters, 6.8±2.1 and 18.0±2.5 mV for AGW and ASW, respectively. Similar stability trends have been observed with regard to simple KCl systems (Chowdhury et al., 2011) and in more complex waters (Keller et al., 2010). Particle size in Fig. 1C for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 1,341.0±95.9, 1,297.0±71.9, 2,644.6±515.2, and 2,793.8±158.0 nm, respectively. Other studies have also reported enhanced metal oxide nanoparticle stability (Fang et al., 2009; French et al., 2009; Chowdhury et al., 2013; Lanphere et al., 2013). The reduced stabilities correlate with the higher removal efficiencies in ASW and AGW at both the flocculation and sedimentation stages and the reduced particle sizes observed in the KCl suspensions, 1,341.0±95.9 and 1,297.0±71.9 nm in 1.83 and 10 mM KCl, respectively, compared with simulated waters, 2,644.6±515.2 and 2,793.8±158.0 nm in AGW and ASW, respectively.
The increase in removal between simulated waters (AGW and ASW) and simple KCl solution is attributed to the presence of divalent cations (i.e., Ca2+, Mg2+) in solution, which reduces particle stability (Benjamin, 2002). Ca2+ ions lead to greater electrical double layer compression than monovalent ions due to the larger outer valence shell size (Gregory, 2006). This phenomenon is most likely due to charge screening and reduced Debye length (Elimelech et al., 1998; Gregory, 2006). As observed in Fig. 1, removal was the greatest (>98%) in AGW and ASW where divalent ions are present. Previous studies have also demonstrated similar effects where calcium ions contributed to enhanced nanoparticle aggregation and larger particle sizes (Chen et al., 2006, 2007; French et al., 2009; Quevedo and Tufenkji, 2012; Torkzaban et al., 2013). Increased hydroxide precipitation is also a likely cause of the improved TiO2 removal due to the presence of highly charged anions, such as sulfate, which promote the hydroxide precipitation that enmesh the smaller TiO2 particles to form significantly larger secondary particles that easily settle out of solution (Duan and Gregory, 2003).
Role of NOM in removal of bare TiO2 nanoparticles
In engineered systems, the presence of NOM is expected to significantly impact the removal and transport of ENMs; therefore, the effect of NOM in all four water sources (1.83 mM KCl, 10 mM KCl, AGW, and ASW) was investigated. A significantly different removal trend was observed compared with the systems without NOM (Effects of IS and Solution Chemistry on Removal of Bare TiO2 Nanoparticles section). Total particle removal in the presence of NOM in Fig. 2A for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 90.8%±0.9%, 23.0%±9.3%, 74.9%±1.1%, and 80.4%±2.6%, respectively. NOM is, in simplest terms, a complex anionic polymer containing carboxylic and phenolic functional groups (Crittenden and Harza, 2005; Viessman, 2009). The presence of ionizable groups within NOM will lead to an expanded form once suspended (and hydrated) due to the repulsion between charged groups. With increasing salt concentration (IS), the repulsion between these groups and other like charged materials in suspension will be diminished (Gregory, 2006). Therefore, more NOM is expected to adsorb onto the TiO2 aggregates at 10 mM KCl as compared with 1.83 mM KCl.

Bare TiO2+SRHA removal was plotted in
Zeta potential in Fig. 2B for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 11.1±2.1, 22.0±3.5, −1.3±3.1, and 17.3±1.9 mV, respectively. The initial charge reversal was observed (zeta potential, Fig. 2B) in the 10 mM KCl system, which is in agreement with the reduced removal observed in Fig. 2A. The significant increase in charge reversal observed between the 1.83 mM KCl system and the 10 mM KCl system may be attributed to the substantial increase in absorbed positively charged hydrolyzed species and hydroxide particles attracted to the negatively charged species within the NOM (Crittenden and Harza, 2005). In contrast, there was no observed statistically significant difference between ASW and AGW, which indicates that sweep flocculation masked the difference between these two systems due to the rapid hydroxide precipitation associated with the highly charge anions.
Overall, there is a subtle reduction in aggregate sizes in the presence of NOM (Fig. 2C) as compared to without (Fig. 1C) NOM, which is attributed to the steric repulsion of the absorbed NOM onto the TiO2 surface. Particle size in Fig. 2C for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 1,054.5±40.5, 1,590.2±150.2, 1,966.1±312.1, and 1,916.8±197.8 nm, respectively. Aggregate sizes increased significantly (p<0.05) with increasing IS within the KCl systems after sedimentation. The aggregate sizes between the simple KCl and simulated systems also significantly increased (p<0.05), although there was insignificant difference in aggregate sizes between AGW and ASW (p>0.05). However, after flash mixing, larger aggregates are observed at 1.83 mM KCl than at 10 mM KCl (Fig. 2C, at 0 min after initial aggregation before settling), which is likely due to the weaker interaction between NOM and TiO2 aggregate surface. Weaker interactions between the NOM and the surface will allow the chain segments to likely extend further into the solution, increasing polymer bridging between particles to form larger aggregates. However, for stronger interactions (10 mM KCl), the NOM conformation will be more condensed, increasing coverage of the aggregate surface, which may increase particle stability due to electrosteric repulsion (Gregory, 2006; Chowdhury et al., 2012). The presence of SRHA has been previously observed as increasing the stability of TiO2 in NaCl and CaCl2 over a range of IS and pH due to electrostatic and steric repulsion (Thio et al., 2011). However, the presence of divalent ions such as calcium in the simulated waters may result in calcium bridging to form much larger aggregates in addition to increased compression of the EDL compared with the simple KCl systems (Chowdhury et al., 2012). This was observed in another study where the deposition kinetics of fullerene (C60) nanoparticles, in the presence of CaCl2, humic acid-coated surfaces increased due to macromolecules undergoing complex formation with calcium ions, reducing the charge and steric influences of the adsorbed macromolecular layers (Chen and Elimelech, 2008).
Role of DMSA coating and NOM on removal of TiO2 nanoparticles
The most environmentally complex yet relevant conditions of this study include the presence of both NOM and a synthetic organic coating representative of commercial coatings used to stabilize such ENM. DMSA was selected as the model synthetic coating, as it is used in several environmental and biomedical applications which would likely have consumer products that may be emitted into such water treatment facilities (Aposhian and Aposhian, 1990; Asiedu et al., 1995). Total removal of DMSA-coated particles for 1.83 mM KCl, 10 mM KCl, ASW, and AGW systems was 85.0%±2.8%, 85.8%±12.0%, 74.9%±1.6%, and 98.5%±0.6%, respectively. The total removal after sedimentation for the KCl systems was statistically insignificant, indicating that in system, monovalent ion system IS had little to no effect similar to the observed results of bare TiO2 due to the strongly adsorbed positively charged hydrolyzed species, resulting in charge reversal and significant removal associated with sweep flocculation. As expected, there was significant charge reversal observed; however, there was no statistically significant difference observed between the zeta potential of 1.83 mM KCl and 10 mM KCl (24.7±2.5 and 25.7±4.2 mV, respectively). Overall, there is a slight reduction associated with the presence of the synthetic organic coating, which is due to the steric repulsion of the organic ligands coating the TiO2, which is also observed in the smaller aggregates observed in DLS (Fig. 3C). There is little significant difference between the KCl systems and the simulated systems for the exception of the increased removal in AGW that is associated with the increased charge screening in the presence of several divalent ions (significant reduction in zeta potential, Fig. 3B) and the promotion of hydroxide precipitation in the presence of strongly charged anions (increased aggregate size, Fig. 3C). The increased removal in the 10 mM KCl system may be related to the chelation of the DSMA between the TiO2 and the rapidly formed hydroxide precipitates (discussed in the earlier sections), reducing the observed stability offered by the presence of NOM. Therefore, the synthetic organic coating that is of significant concern for environmental and engineered systems has minimal impact on the behavior compared with the behavior of the ENMs without any synthetic coating.
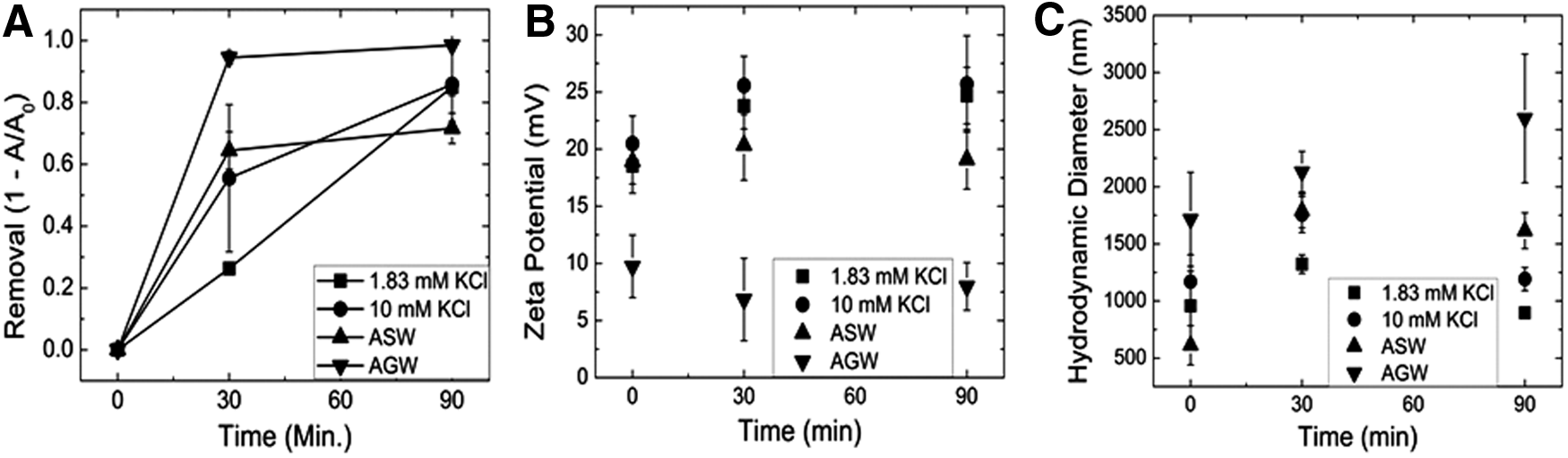
DMSA-coated TiO2 removal was plotted in
Although the scenario of most significant concern is the impact of NOM on coated nanoparticles, this is the most probable state that such ENMs will exist within engineered water treatment systems. Total coated particle and NOM removal in Fig. 4A for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 89.1%±5.0%, 84.9%±4.0%, 71.8%±3.0%, and 68.7%±3.4%, respectively. Zeta potential in Fig. 4B for 1.83 mM KCl, 10 mM KCl, AGW, and ASW was 8.2±3.2, 5.6±6.0, −3.1±5.6, and 16.5±2.5 mV, respectively. Interestingly, the removal efficiencies between the simple KCl and the complete simulated systems (AGW and ASW) are inverted from what would be expected. This is related to the observed removal trend of bare TiO2 particles in the presence of NOM. The presence of the coating may strengthen the interaction between the NOM surface, which will reduce the extension of chain segments into solution that would otherwise promote aggregation (Duan and Gregory, 2003; Gregory, 2006). Subsequently, in the simulated waters where charge screening will reduce the charge of repulsion felt between the ionized species within the NOM chains increase the interaction between the surface and the NOM, further increasing the stability associated with steric interactions (Duan and Gregory, 2003; Gregory, 2006). The reduced removal in the simulated waters as compared with the previous scenario with only NOM may be caused by possible chelation of DMSA and other metal ions such as calcium or magnesium, reducing the amount available for bridging (Flora and Tandon, 1990) This is supported by the significantly larger aggregates observed (Fig. 4C) in AGW where there would be more calcium available compared with the other three systems.
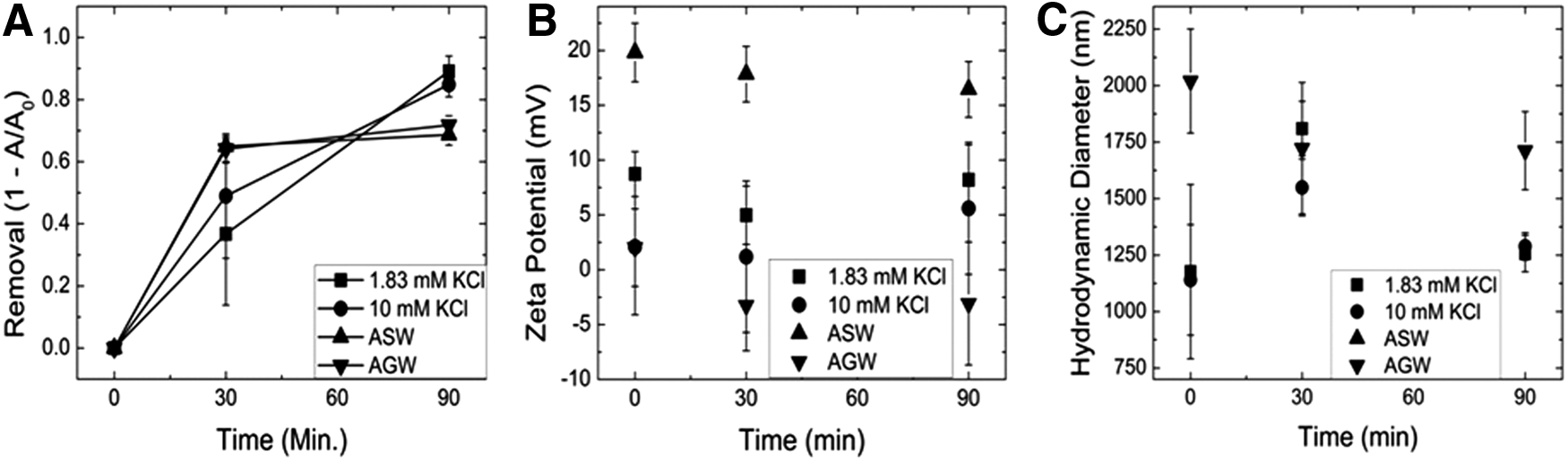
DMSA-coated TiO2+SRHA removal was plotted in
Under all four scenarios investigated, the concentration of TiO2 aggregate particles, smaller than 0.45 μm, was measured using inductively coupled plasma mass spectrometry (ICP-MS) (Table 2). Even in conditions with greater than 90% overall removal, ∼5 ppm of the smaller TiO2 aggregates stayed suspended in the solution. This indicates that regardless of the addition of coagulants and other aggregation inducing compounds, there is still significant concern of the released ENMs in small aggregates via conventional water treatment processes. These are the particles that are of greater health and environmental concern due to their increased reactivity and possible transport through the environment (Gurr et al., 2005; Long et al., 2006; Warheit et al., 2006; Chowdhury et al., 2012; Jassby et al., 2012; Vicario-Parés et al., 2014).
Experimental conditions and parameters are listed in the table.
AGW, artificial groundwater; ASW, artificial surface water; DMSA, meso-2,3-dimercaptosuccinic acid nanoparticle (NP) coating; ICP, inductively couple plasma; ICP-MS, inductively coupled plasma mass spectrometry; NOM, natural organic matter at a constant concentration of 1 mg/L.
Environmental implications
While it was determined that IS, solution chemistry, and water type impacted the removal of TiO2, the impact of environmentally relevant factors such as the presence of humic acids and synthetic organic coatings on the particle coating had a more significant impact on the removal. The main governing removal mechanism as expected was sweep flocculation specifically in the simulated waters (ASW and AGW) due to the presence of highly charged anions (Duan and Gregory, 2003). There was a notable impact on removal in the simple (KCl) systems with the addition of NOM, which is attributed to the degree to which the NOM is associated with the particle subsequent electrosteric repulsion stabilizing the particles (Gregory, 2006). In the simulated waters, there is a less significant deference between the two waters related to the degree of association between NOM and the surface due to the increase in charge screening from the presence of divalent ions; therefore, the hydroxide precipitation (sweep flocculation) dominates, and significant removal is observed (Gregory, 2006). In addition, these divalent ions such as calcium may form bridges increasing the aggregates sizes and therefore the removal of the particles. The presence of a synthetic organic coating on such ENMs is of significant environmental concern due to the potential increase in toxicity associated with the coating (Vicario-Parés et al., 2014); however, the model organic coating had little significant impact on the removal as compared with the bare TiO2 in the first scenario. The most relevant and complex system is the synthetically coating TiO2 (DMSA) in the presence of NOM. An observed inversion of the expect outcome, as observed in the system with just NOM, is observed in this scenario. The presence of the DMSA possibly results in an increased affinity of the NOM to the particle surface and increasing electrostatic repulsion. DMSA may also chelate with the divalent ions in solution, which would reduce the opportunity for calcium bridging (Flora and Tandon, 1990).
It may be expected due to the typically high dosages of coagulants used in practical water treatment that ENMS particles would be near completely removed via rapid precipitation hydroxide particles, which enmesh such particles, during sweep flocculation (Elimelech et al., 1998; Duan and Gregory, 2003). In the previous study by Honda et al., the coagulant type, coagulant concentration, and TiO2 concentration were investigated and optimized under simple conditions. Expanding on this previous study, we investigated realistic water chemistries (i.e., simulated water formulas and addition of NOM) and more relevant materials (ENMs with a synthetic coating). While further optimization of operating conditions such as coagulant concentration and pH may provide further insight and improve removal efficiencies, this study shows that there may be significant release of the materials. Specifically the nearly ∼5 ppm concentration of TiO2 aggregates, observed in all scenarios, smaller than 450 nm that may be released into our surface waters or our distribution systems from these engineered treatment facilities raises the question regarding the efficacy of our current treatment facilities to adequately remove ENMs such as TiO2 and the growing array of new ENMs.
Footnotes
Acknowledgments
This study was supported by the National Science Foundation under Grant No. CBET-0954130. Any conclusions, findings, recommendations, or suggestions expressed in this research are those of the authors and may not reflect the views of the National Science Foundation. N. Kinsinger was supported by the United States Department of Agriculture's NIFA Postdoctoral Fellowship Program. The authors would like to acknowledge Dr. Mark Matsumoto from the University of California, Riverside for providing helpful insights into operating conditions and relevant parameters in the jar tests. In addition, they would like to thank Dr. Haizhou Liu and his graduate student, Lucy Liu, for their help with conducting ICP-MS experiments.
Author Disclosure Statement
No competing financial interests exist.