
Abstract
Abstract
Photocatalytic reduction of hexavalent chromium [Cr(VI)] was investigated to evaluate effectiveness for removing all chromium species from drinking and industrial waters. Deionized and tap water experiments were performed using a system that recirculates TiO2 through an integrated process consisting of ultraviolet (UV) lamps and a ceramic membrane. Hexavalent and total chromium concentrations were simultaneously reduced during treatment. Cr(VI) removal gradually increased with higher energy input and TiO2 dosage, achieving greater than 90% removal for a 1 g/L dose of TiO2. Cr(VI) was photochemically reduced to Cr(III) on the surface of TiO2, where the Cr persisted as a precipitate. Upon further irradiation, Cr(III) could be reoxidized to Cr(VI). High-volume flow-through experiments indicated significantly diminished chromium removal due to catalyst fouling during continuous catalyst use. To greater extents in tap water than in DI water, photoaggregation of the TiO2 catalyst was evidenced by increased particle size. This photoaggregation effect was further supported by decreased breakthrough of TiO2 with increased irradiation intensity.
Introduction
S
Both hexavalent and trivalent chromium [Cr(III)] forms have been linked to adverse health effects, including, but not limited to, increasing risk for cancers (respiratory, prostate, lymphoma, leukemia, bone, and stomach); gastrointestinal system disruption; uptake, accumulation, and toxicity in vital organs; damage to DNA; and gene mutation (Costa, 1997; Dayan and Paine, 2001; Sedman et al., 2006; Beaumont et al., 2008). These findings have raised a concern among the general public and spurred a campaign to regulate Cr(VI) and decrease its level in drinking water. With California leading the way and setting an enforceable maximum contaminant level for hexavalent chromium at 10 μg/L, and in expectation of a major effort led by the EPA to promulgate a similar national hexavalent chromium standard, many utilities are exploring treatment options to address these upcoming regulatory requirements (California Environmental Protection Agency, 2011; Barrera-Díaz et al., 2012; Gore, 2014).
Treatment options for Cr(VI) have traditionally fallen into six categories (Sharma et al., 2008): (1) coagulation–precipitation–filtration, (2) adsorption to different media, (3) ion exchange, (4) membrane technology, (5) electrodialysis, and (6) biological removal. Challenges with these technologies include cost, scalability, and reliability to achieve low Cr(VI) concentrations (Owlad et al., 2009; McNeill et al., 2012). Hexavalent chromium is an oxyanion that adsorbs poorly to most metal oxides at neutral pH ranges (pH=7±1) because of the repelling forces generated by the negatively charged surface of the metal oxide and the anionic chromate/dichromate forms. Ion exchange technologies generate brines containing high concentrations of hexavalent chromium, while the other technologies are either uneconomical or cannot remove sufficient hexavalent chromium to achieve anticipated regulatory limits. In contrast, water treatment technologies based on photocatalytic reduction processes are able to overcome these challenges by reducing hexavalent chromium to a trivalent form, reversing the charge on the chromium species and inducing favorable sorption to metal oxide surfaces, which simultaneously may serve as photocatalysts.
A number of studies have demonstrated that uniquely synthesized and modified semiconductor ultraviolet and visible (UV/VIS) light active photocatalysts are capable of reducing and removing hexavalent chromium from water to concentrations anticipated in the upcoming regulations (Chakrabarti et al., 2009; Li et al., 2012; Vignesh et al., 2013; Yu et al., 2015). However, much of the documented work focuses on laboratory-scale conditions and commercially unavailable photocatalysts (Padhi et al., 2014; Zhao et al., 2014; Li et al., 2015) and, as such, it is not readily translatable to full-scale commercial applications (Liu et al., 2014).
The existing literature suggests that titanium dioxide (TiO2) may be among the few inexpensive and commercially available photocatalysts capable of addressing the majority of these deficiencies (Doudrick et al., 2012; Ghorab et al., 2013). Although titanium dioxide is conventionally viewed as a photocatalyst suitable for full-scale, advanced photo-oxidation processes because of its hydroxyl radical generation capacity (i.e., oxidation), properly designed experimental conditions also allow for successful utilization of its photoinduced reduction capabilities. The capability for TiO2 photoinduced reduction of hexavalent chromium has been demonstrated in a limited number of laboratory-scale studies (Gimenez et al., 1996; Chen and Ray, 2001; Ku and Jung, 2001; Wang et al., 2008; Yang et al., 2012); the photocatalytic reduction properties of TiO2 for removing hexavalent chromium in a full-scale commercially available reactor had not been explored before this study. The overarching goal of this study was to examine the feasibility of using a commercially available integrated UV reactor system with commercial grade titanium dioxide to reduce and remove hexavalent chromium from water.
To achieve the goal, five objectives were assessed: (1) the efficacy of the commercially available photocatalytic system for reduction of hexavalent and total chromium was verified in ultrapure water to exclude potentially interfering species; (2) the influence of water matrix effects on contaminant removal was examined in tap water and buffered deionized water to assess system performance under realistic conditions and to evaluate potential for scale-up; (3) oxidation of trivalent chromium adsorbed on titanium dioxide surfaces to hexavalent chromium and subsequent suspension upon further irradiation was investigated to quantify potential for back reaction within the photocatalytic system; (4) surface analysis of the titanium dioxide catalyst for the presence of chromium on the titanium dioxide surface and photoinduced aggregation of the catalyst was conducted to validate removal mechanisms and investigate potential limitations of continuous catalyst use without regeneration; and (5) the potential for catalyst leaching was investigated through analysis of membrane permeate to determine a realistic catalyst loss scenario. These objectives address knowledge deficiencies for pilot-scale application of photocatalytic reduction of hexavalent chromium and provide the first insights into the scalability of this technology beyond the laboratory scale.
Methodology
Photocatalytic efficacy of a commercially available system for reduction of hexavalent and total chromium
Based on previously published work, which demonstrated that commercially available photocatalytic systems can successfully oxidize organic compounds (Benotti et al., 2009; Westerhoff et al., 2009) and disinfect water (Gerrity et al., 2008), the Photo-Cat® Serial 0700 system (Purifics ES, Inc.) was selected to investigate the photocatalytic reduction capabilities of such systems for treatment of hexavalent chromium. The Photo-Cat® Serial 0700 system is an integrated UV/ceramic membrane reactor containing four 220 W low-pressure mercury UV lamps controlled by an automated process control system (Fig. 1). These low-pressure lamps emit UV light with λ=253.7 nm, which is sufficient to activate the TiO2 band gap 3.2 eV (Doudrick et al., 2012) and create hole/electron pairs. Unmodified Evonik P90 TiO2, which has same crystallinity as P25, but smaller crystal particle size and higher surface area, was used as a TiO2 photocatalyst to minimize the electron/hole recombination effect (Doudrick et al., 2012, 2013). The Photo-Cat system allows for complete recovery and reutilization of the TiO2 photocatalyst by recirculating it through the ceramic ultrafiltration membrane. In addition to operating in a recirculation mode only (i.e., no active lamps), the Photo-Cat system allows for variable power output control by operating one or multiple lamps simultaneously.
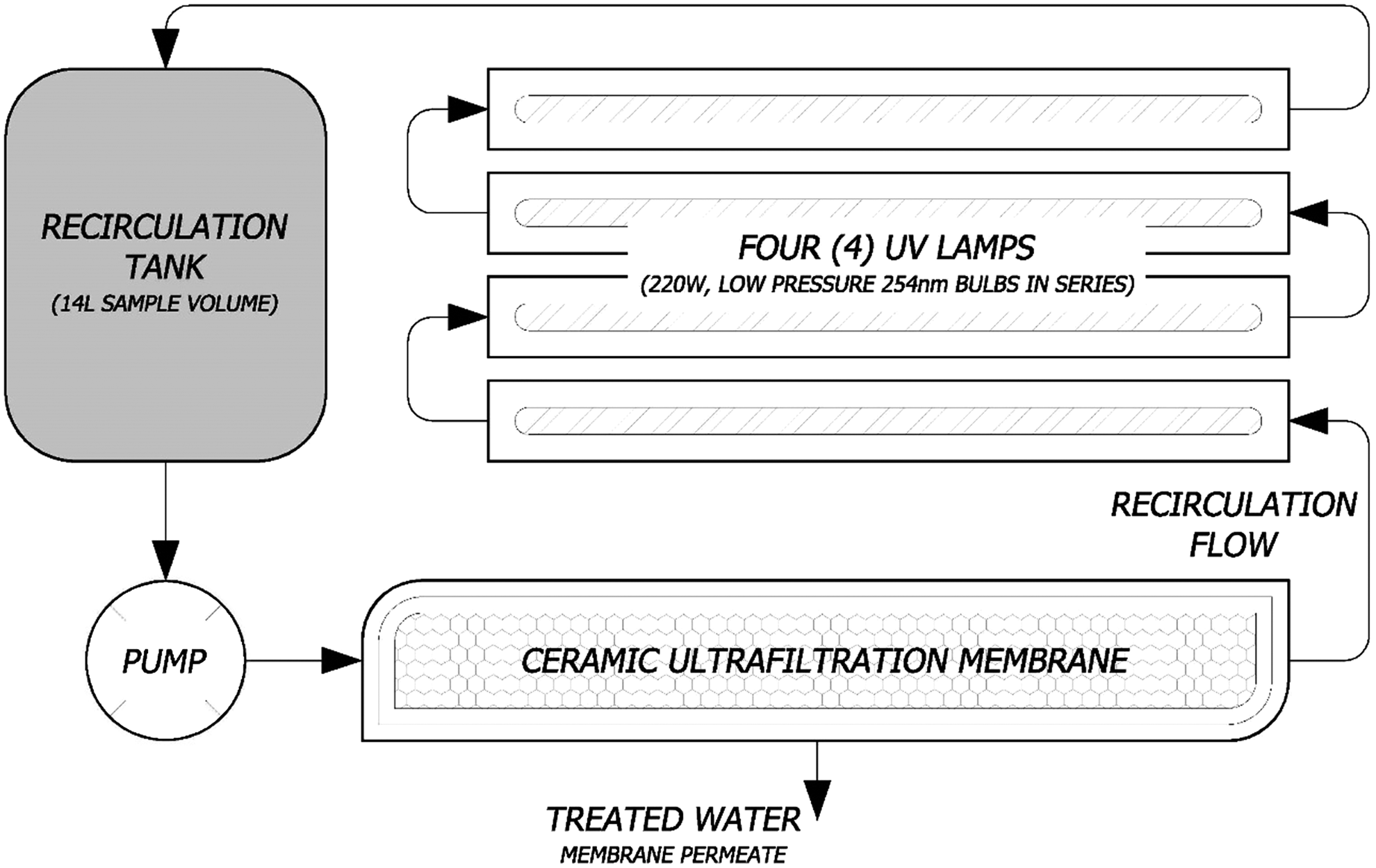
Schematic of pilot-scale photocatalytic reactor, Photo-Cat L®, by Purifics.
Initial photocatalysis experiments were conducted in ultrapure water (<1.5 μS/cm, >18.3 MΩ·cm) to verify the system's ability to remove chromium and exclude the potential interference of species that could impair the photocatalytic reduction process. The experimental matrix examined the hexavalent chromium reduction performance of the system at different contaminant/catalyst ratios, which included two initial hexavalent chromium concentrations (100 and 500 μg/L) and three TiO2 doses (0.01, 0.1, and 1.0 g/L P90). Although these hexavalent chromium concentrations generally exceed values found in natural systems (Seidel and Corwin, 2013), high concentrations were used to ensure observable concentration changes throughout the experiments. Potassium dichromate (K2Cr2O7, >99%; Sigma Aldrich) was used as the source of the hexavalent chromium.
Experiments were conducted using the Photo-Cat system's recirculation mode at a programmed flow rate of 20 L/min and at UV lamp energies ranging from 0 to 8 kWh/m3 (calculated based on run time to normalize results to other commercial systems) with total treatment volume of 14 L. Temperature of the system was maintained at 27.5±2.5°C by running cooling water across the lamps. Control experiments with no TiO2 were also conducted to assess the reduction capability of the system without the presence of TiO2 through photolysis. External, organic hole scavengers and pH control were not used during experiments. During the experiments, the pH ranged between 6.5 and 7.1. The system was purged with a minimum of 250 L of deionized water between experiments to eliminate potential for crossover contamination.
As part of the quality control process, lamp performance was assessed periodically by conducting methylene blue photodegradation tests. These tests ensured comparable performance for all lamps. Electrical energy per order (EE/O) was calculated for photocatalytic batch experiments following the method described by Bolton and Stefan (2002). Separate experiments were performed in a flow-through operation using a feed of dechlorinated tap water (pH ∼7.5) spiked with either 10 or 100 μg/L Cr(VI). The steady-state flow rate through the system was 2.1 L/min. Lamps were operated at full power in the presence of 1 g/L TiO2, which was captured by the ceramic membrane, recirculated, and mixed with feed water. In each experiment, a fully mixed system and period of dark adsorption were completed before illumination. Thus, the system was at a steady state (pH, well mixed, dark adsorbed, temperature) before illumination.
Sample aliquots (40 mL) were taken at regular time intervals from the system's effluent port located after the ceramic membrane. The aliquots were additionally filtered through 0.45-μm filters to ensure the absence of any aggregated TiO2 nanoparticles. Samples were acidified (1) with ultrapure nitric acid for analysis through inductively coupled plasma mass spectrometry (ICP-MS) or (2) with ammonium hydroxide buffer solution for ion chromatography. Modified EPA method 218.6 was used to determine hexavalent chromium concentrations by ion chromatography (Dionex ICS 2000) following a manufacturer-recommended, post-column derivatization method with 1,5-diphenylcarbazide and ammonium sulfate eluent. Total chromium concentrations were measured using a quadrupole ICP-MS (Thermo Fisher Scientific XSeries 2).
Experiments in buffered deionized, pH-adjusted deionized, and tap water
To examine the influence of water matrix on contaminant removal and assess the system's performance, water matrices with increasing complexity were used: (1) 5 mM NaHCO3 buffered ultrapure water, (2) deionized water with added potassium hydroxide to adjust pH without adding carbonate alkalinity, and (3) a more complex tap water matrix (dechlorinated tap water from the City of Tempe, Arizona, distribution system with hardness=220 mg/L as CaCO3, TDS=615 mg/L, and conductivity 1.0±0.1 mS/cm) (City of Tempe, 2012). The pH of the 5 mM NaHCO3-buffered ultrapure water was 8.6±0.1, and the pH of the tap water was 7.8±0.1. The pH of the KOH-adjusted solution varied from 6.5±0.1 to 9.0±0.1. Water samples were prepared by spiking 500 μg Cr/L hexavalent chromium (potassium dichromate, K2Cr2O7, >99%; Sigma Aldrich) into the various water matrices. Input lamp energies ranged from 0 to 31 kWh/m3. Experimental protocol was identical to that followed for the ultrapure experiments above.
Quantifying potential for oxidizing photocatalytically reduced Cr(III) to Cr(VI)
As titanium dioxide photocatalysis is inherently an oxidation-reduction system, the potential for oxidation of photocatalytically reduced trivalent chromium sorbed onto the titanium dioxide surface was quantified to determine potential for resuspension of hexavalent chromium upon excess irradiation. Chromium (III) chloride hexahydrate (CrCl3·6H2O, >98%; Sigma-Aldrich) was used as the source of trivalent chromium and was spiked into the system to achieve dosages of 100 and 500 μg-Cr/L. Identical protocols were followed to previous photocatalytic experiments at catalyst dosages of 0.1 and 1.0 g/L P90.
Characterization of spent TiO2 photocatalyst
Spent TiO2 photocatalyst samples from all three water matrices were examined to determine any photocatalyst poisoning or sorption of chromium onto the P90. The samples were dried at 100°C on an aluminum stub and left to equilibrate at room temperature (22°C) before electron microscopy analysis. Scanning electron microscopy equipped with an energy dispersive X-ray microanalysis system (SEM/EDX; Philips XL30-EDAX) was used to determine the presence of chromium on the surface of the titanium dioxide photocatalyst.
Occurrence of TiO2 photocatalyst nanoparticles in treated effluent
Samples for aqueous titanium analysis were collected from the ceramic membrane permeate sample port (Fig. 1) to determine potential for nanoparticle leaching from the reactor. Single particle ICP-MS (spICP-MS), an emerging nanoparticle quantification and size characterization technique (Degueldre et al., 2006; Mitrano et al., 2012), was used to evaluate the particulate TiO2 equivalents in the photocatalytic reactor effluent. Effluent samples were introduced directly into the ICP-MS, and the instrument signal in counts per second (cps) was documented over time. Dwell time, the unit time interval in which one reading was integrated, was set as 10 ms, and the sample flow rate was set as 0.69 mL/min. Nebulizer transport efficiency to be used in Ti quantification was determined as 1.58% based on previous research (Pace et al., 2011). Total Ti concentration was evaluated by considering the elevated baseline relative to the blank and counting the pulses that stand for the detectable particle signals.
Results and Discussion
Cr(VI) removal from ultrapure water
Cr(VI) concentrations slowly decreased by direct UV photolysis in ultrapure water without TiO2 (Fig. 2). Initial concentrations appear to vary, but reflect different amounts of dark adsorption for the varied catalyst doses. A 1 g/L TiO2 dosage had the highest rate and most complete removal of Cr(VI), achieving nondetect levels. An unexpected finding was that photolysis (no added TiO2) was marginally more effective than adding a very low dosage of 0.01 g/L. It is likely that the low TiO2 dosages reduced UV transmittance, thus limiting direct photolysis of Cr(VI) while providing minimal surface on the TiO2 for electron transfer to Cr(VI). The highest titanium dioxide dose (1.0 g/L TiO2) had an EE/O value of 0.36 kWh/m3, which is within the cost-effective range (Crittenden, 2012). The EE/O for 0.1 g/L TiO2 was found to be an order of magnitude higher (8.3 kWh/m3). Based on these findings, two TiO2 dosages (0.1 and 1.0 g/L) were tested for four water matrices and under different irradiance conditions.
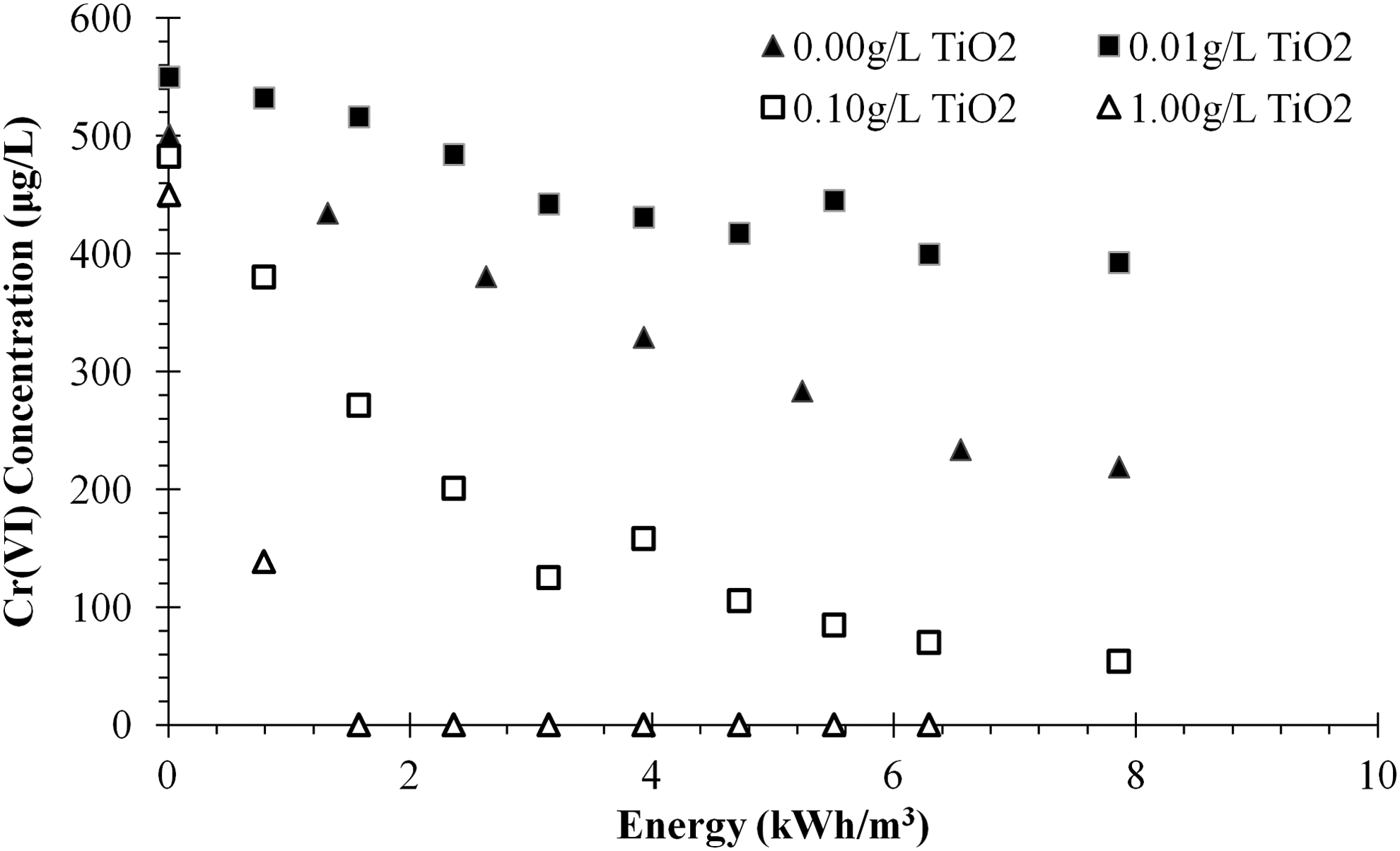
Hexavalent chromium removal as a function of energy using four TiO2 catalyst dosages in model water matrix (18.3 MΩ nanopure deionized water), with one of four operational lamps running in recirculation mode. pH ranged from 6.5 to 7.1 (initial to final), and temperature was maintained between 25°C and 30°C.
Cr(VI) removal from complex water matrices
In recirculation experiments, hexavalent and total chromium concentrations decreased simultaneously for a dechlorinated tap water matrix spiked with different initial Cr(VI) concentrations. This indicates that both hexavalent and total chromium were removed from the aqueous solution (Fig. 3). Using 1 g/L TiO2, chromium removal varied between 89% and 98% for initial concentrations of 500 μg/L Cr(VI). Only 4% of Cr(VI) adsorbed onto TiO2 in the dark. Therefore, Cr(VI) removal greater than 4% is attributable to photocatalytic processes. With only 0.1 g/L TiO2 and 100 μg/L Cr(VI), removal varied from 45% to 70% with ∼6.5% dark adsorption. The change in Cr(VI) concentration equaled the removal of total chromium in the system, indicating sorption of chromium species onto titanium (discussed below). EE/O for dechlorinated tap water was substantially (∼10×) higher than ultrapure water resulting from catalyst fouling and diminished availability of sites for adsorption and reduction.
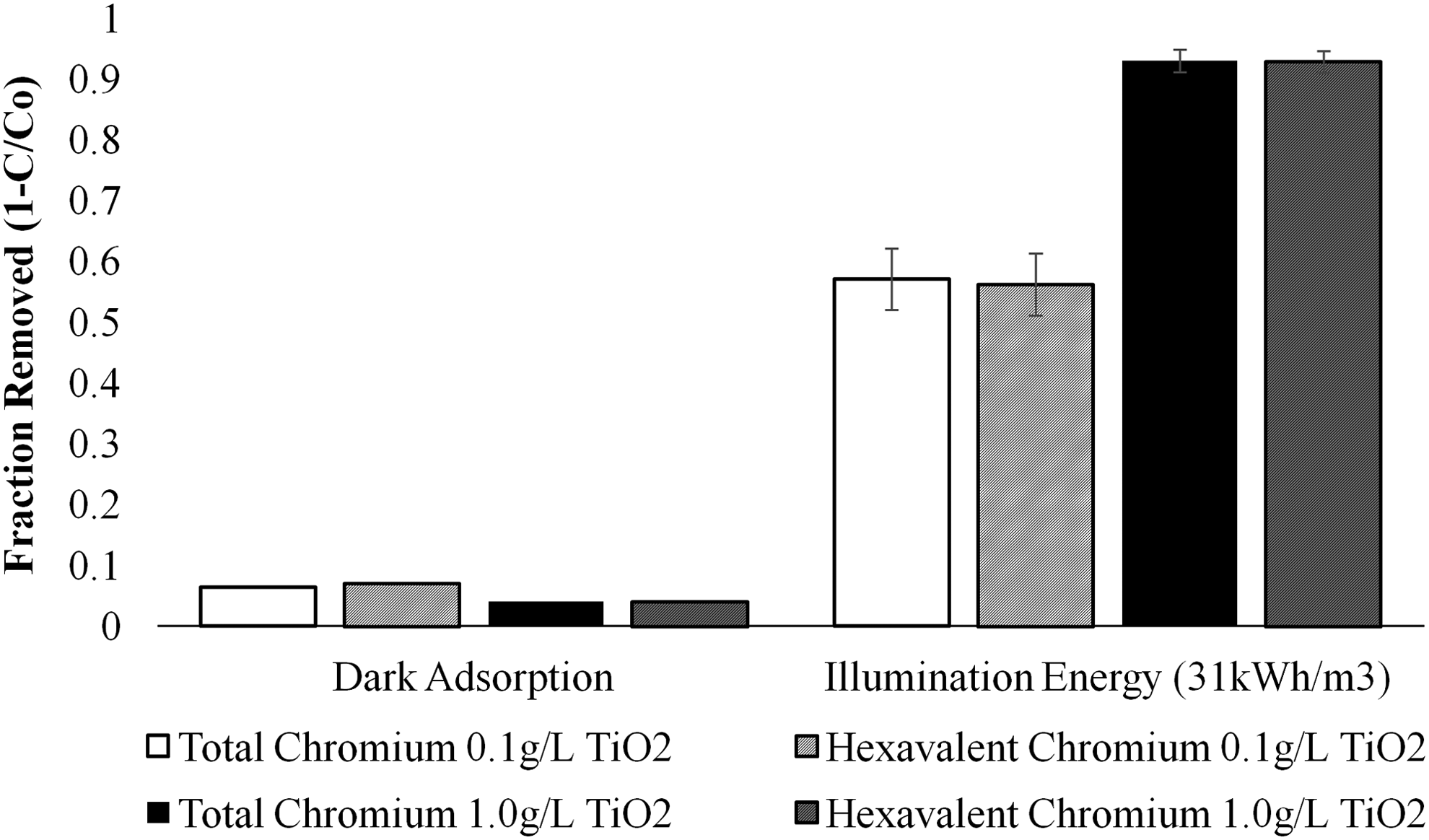
Removal efficiencies for hexavalent and total chromium in dechlorinated tap water with initial Cr(VI) concentration of 500 μg/L. The y-axis represents calculated removal of either hexavalent (striped) or total chromium (solid), while the x-axis represents the energy input. Dark adsorption was measured after a runtime of 1 h without illumination. Illumination data sets show removal normalized to an equivalent energy input (31 kWh/m3) with error bars (n=1/2).
The impact of water matrix was assessed using four water sources: 18.3 MΩ·cm nanopure water, buffered deionized water (5 mM NaHCO3), pH-mediated deionized water with KOH, and dechlorinated tap water. In ultrapure water with only Cr(VI), >99% Cr(VI) reduction occurred with 2 kWh/m3 of energy input (Figs. 1 and 4). In 5 mM NaHCO3-buffered deionized water, TiO2 dosages below 1 gTiO2/L achieved less than 20% Cr(VI) removal, regardless of energy input, and a 1 g/L dose of TiO2 achieved 50% reduction of 500 μg/L Cr(VI) at 10 kWh/m3. Dechlorinated tap water exhibited slightly more effective Cr(VI) reduction than 5 mM NaHCO3-buffered deionized water, with removal highest at 62% with a 1 g/L TiO2 dose.
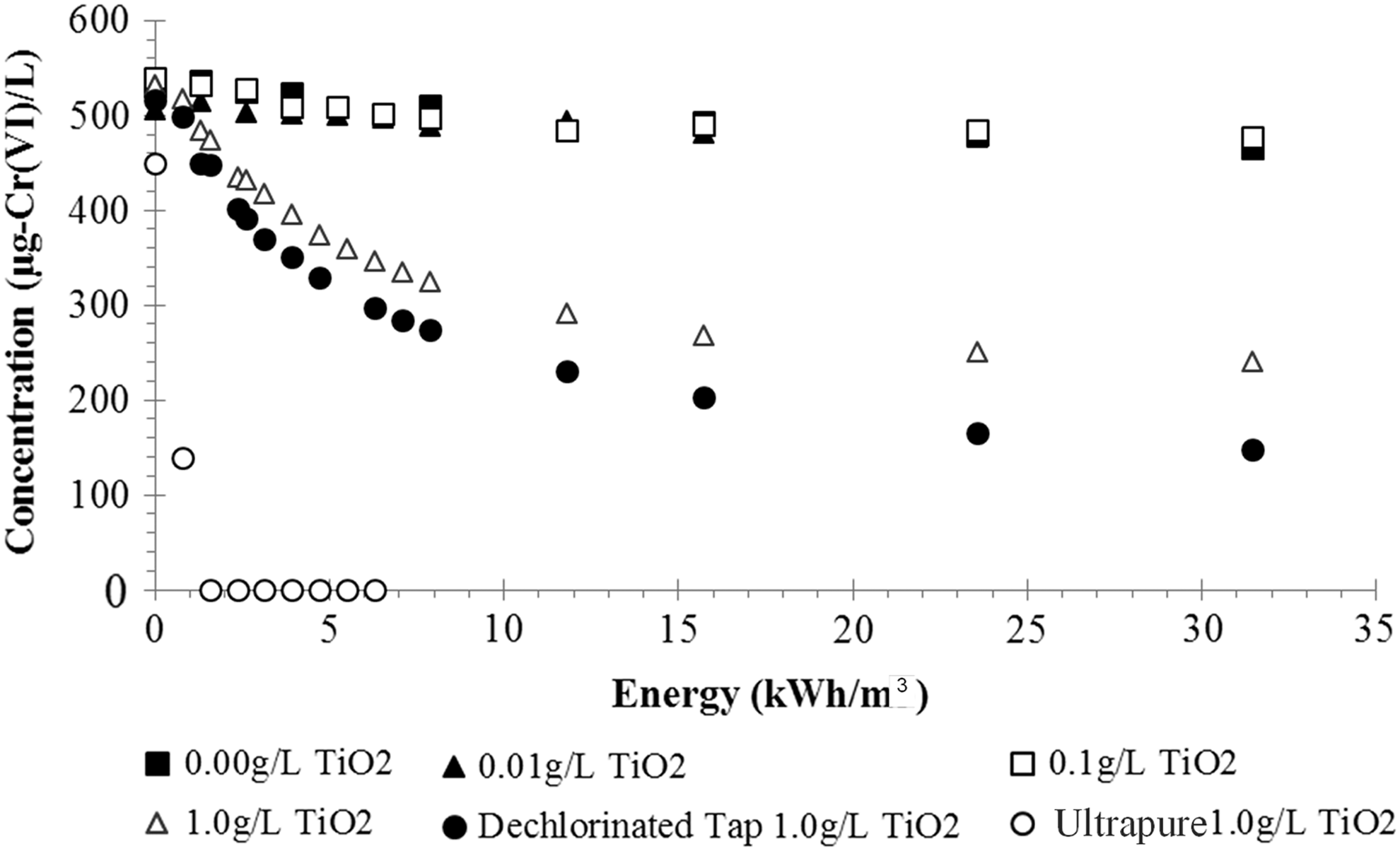
Effluent chromium concentrations based on initial input of P90 dosage. Initial Cr(VI) concentration was 500 μg/L, and P90 titanium dioxide was 0.0, 0.01, 0.1, or 1.0 g/L. Experiments were conducted using buffered deionized water (5 mM NaHCO3, pH 8.5–8.7) unless otherwise noted in the legend. pH for dechlorinated tap matrix ranged from 7.7 to 7.9 from Cin (at t=0) to Cf (final sampling); pH for ultrapure ranged from 6.5 to 7.1. Temperature was controlled to remain between 25°C and 30°C for all experiments.
Chromium hydroxide passivization of the surface may have been a predominant factor in the saturation observed in the chromium reduction of Figure 4. Additionally, at higher pH, the initial likelihood of Cr(VI) adsorption to the surface diminishes, and pH was observed to increase throughout experiments due to the reduction processes and lack of added hole scavenger. Because 5 mM NaHCO3 increases pH by one unit, which creates less favorable thermodynamic conditions for Cr(VI) reduction, further experiments were completed using deionized water without a buffer (Fig. 5). A significant dark adsorption of chromium (CrO42−, pKa=0.6 and 5.9 from Brito et al., 1997) occurred when the solution pH was within±1.5 pH units of pHzpc for TiO2 (pHzpc=6.2). Desorption occurred upon initial irradiation, followed by reduction to Cr(III) upon further irradiation, sorption, and thus removal from the aqueous phase. When the pH of the solution was greater than 7.8 (pH >1.5+pHzpc), less dark adsorption of chromium species was observed. With multivalent cations [i.e., Cr(III)], small changes in pH can lead to relatively large changes in sorption capacity, as evidenced in Figure 5.
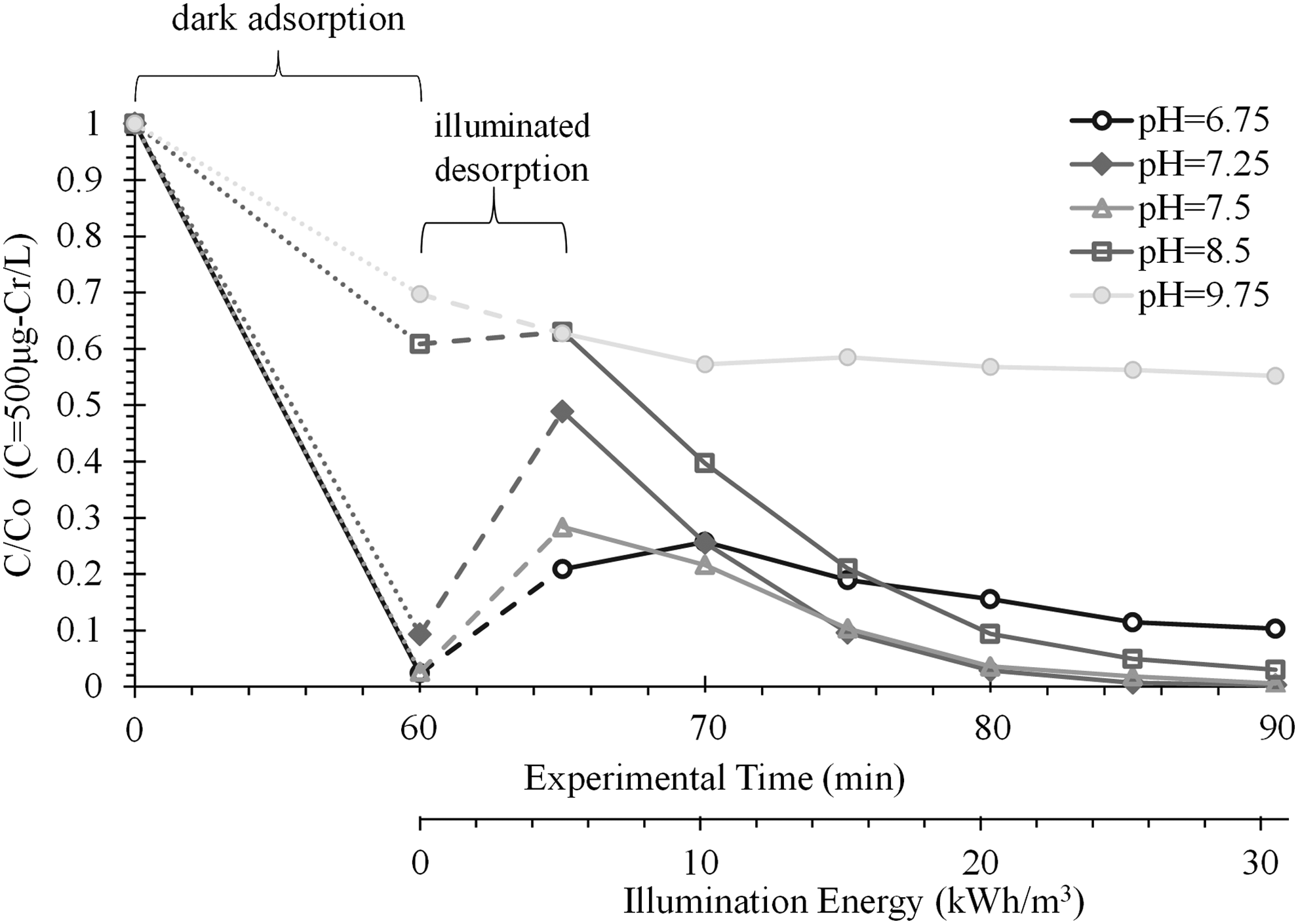
Comparison of chromium removal at varied pH for a deionized matrix. Initial Cr(VI) concentration (C0) was 500 μg Cr(VI)/L. pH was varied using aliquots of KOH solution and equilibrated for 60 min before irradiation. Secondary x-axis corresponds to the illumination energy upon irradiation (beginning at t=60 min).
Spent TiO2 surface analyses
Analyses conducted on slurry effluent samples taken after dechlorinated tap experiments showed accumulation of Cr on the TiO2 solid surface. While virgin P90 TiO2 is pure white, the dried titanium samples from experiments were green, an indicator of chromium species on the surface of the titanium dioxide. SEM was conducted on both virgin P90 and a Photo-Cat slurry effluent from a dechlorinated tap water experiment series of 1 g/L P90 and 500 μg/L Cr(VI) to determine the presence and quantity of chromium on the surface (Fig. 6). For the case of virgin P90, SEM-EDX showed only Ti and O present at the surface, with a carbon response from the stub on which the P90 was mounted. SEM-EDX analysis on spent TiO2 indicated chromium in addition to a number of common tap water constituents (based on EDX: Na>Cl>Mg>Cr>S>Ca>K) on the TiO2. SEM of TiO2 after flow-through experiments revealed a doubling in size of TiO2 particles compared with the batch mode, but with comparable distribution of elements on the catalyst surface. Although chromium represented a relatively low-atomic-weight percentage of surface coverage, detection shows that it was present and attached to the surface in some manner.
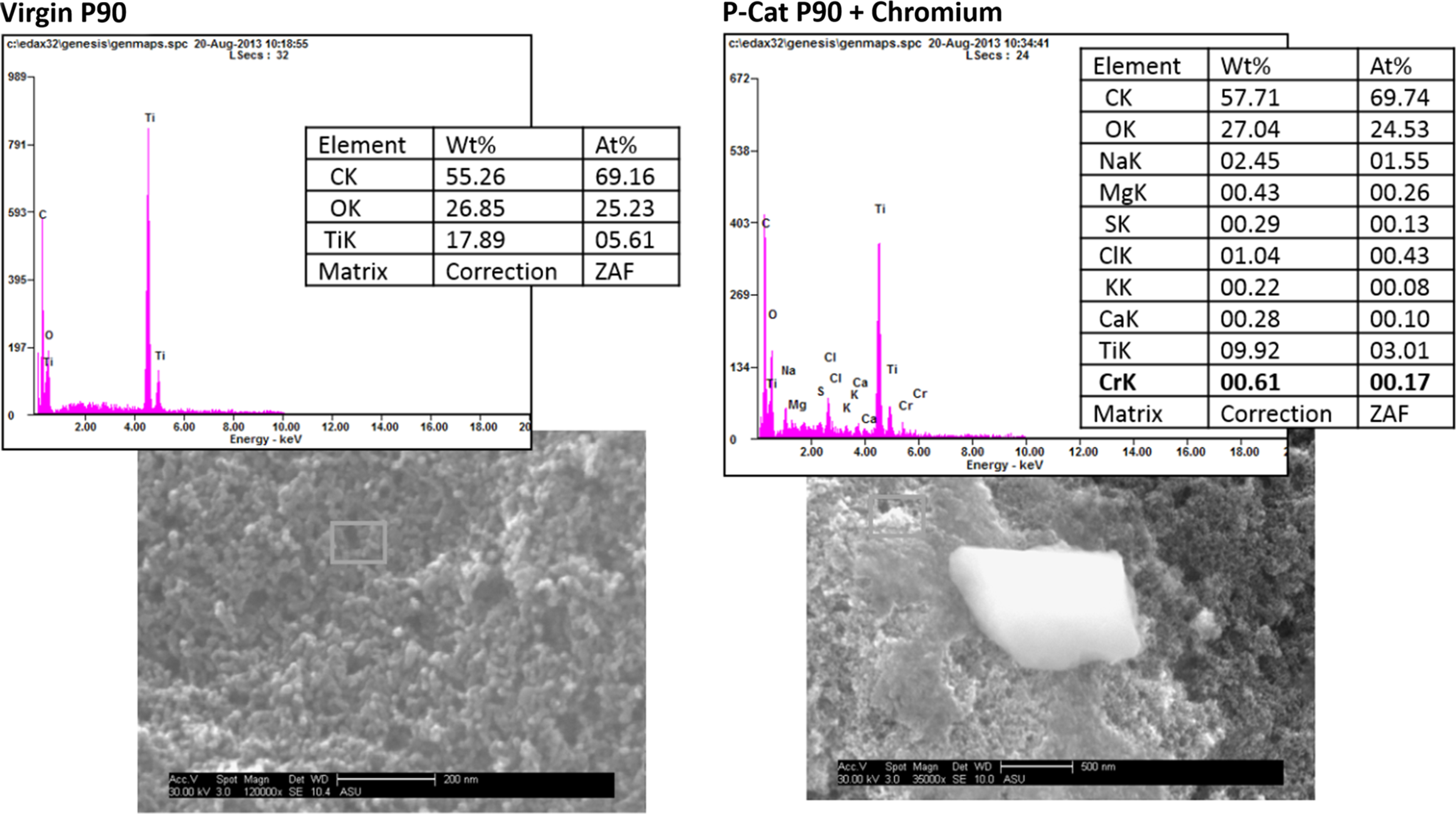
SEM images of virgin P90 versus P90 run in Photo-cat with 500 μg-Cr/L and 1 g-P90/L.
Cr(VI) reformation for tap water spiked with Cr(III)
Figure 3 shows that in the recirculation operation mode, a steady-state concentration of Cr(VI) and an equal concentration of soluble Crtotal (total chromium) occurs in solution. After Cr(VI) concentrations in the ceramic membrane permeate water decreased to below detection levels, continued and prolonged UV irradiation and photocatalysis may reform Cr(VI). This likely occurred as Cr(III) bound to the surface of the TiO2 was oxidized to Cr(VI). Cr(VI) reformation potential was investigated using an initial input of 100 μg/L Cr(III) with 0.1 g/L P90 and 500 μg/L Cr(III) with 1.0 g/L P90. Figure 7 shows that in both cases, Cr(VI) evolved from the Cr(III) initial solution. Less than 10% of the Cr(III) became soluble Cr(VI) at 0.1 g TiO2/L and 100 μg Cr/L, and even less (<2%) formed at the higher TiO2 and Cr(III) doses. This may be due to both the high sorption of Cr(III) to TiO2 and precipitation of Cr(OH)3(s) at neutral pH. The conceptual model presented in Figure 8 articulates the sorption–desorption–reduction cycle with recommendation of a mid-process recycle for the spent TiO2.

Evolution of Cr(VI) from a starting concentration of only Cr(III) in dechlorinated tap water. pH increased over the course of the experiment (7.5–7.75 and 7.85–7.95 for 1.0 and 0.1 g/L TiO2, respectively).

Conceptualization of mechanisms involved in reduction of hexavalent chromium and removal from aqueous solution of total chromium species through reduction and sorption processes. Boxed items represent sorbed species. The phases indicated represent (I) Cr(VI) reduction to Cr(III) and sorption to TiO2 surface; (II) Cr(III) stability on the surface and opportunity to recycle titanium and remove chromium species using acid rinse; and (III) Cr(III) oxidation and desorption upon additional irradiation.
For 10 and 100 μg/L influent Cr(VI) flow-through experiments, effluent measurements of Cr(VI) and Crtotal were indifferent and averaged 9.4±0.7 and 79.8±1.8 μg Cr/L after 30 min, respectively. These experiments suggest either poor photocatalytic reduction of Cr(VI) in the tap water or steady-state removal and reformation. Poor photocatalytic reduction is suspected because evaluation of spent TiO2 indicated significant aggregation of the media, which would reduce light exposure on the surfaces and potentially reduce ability of Cr(VI) to directly contact the TiO2. Figure 6 shows SEM images of virgin TiO2 and spent media from continuous flow tap water experiments. EDX analysis of the latter media indicated the presence of Na, Cl, Mg, Cr, S, Ca, and K. In contrast to a limited concentration of natural water foulants in batch experiments with tap water, continuous loading of foulants from tap water appears to have caused aggregation of TiO2 and reduction in chromium removal efficiency. Prior research has suggested that fouling of TiO2 reduces effectiveness of the Photo-Cat system during the long-term operation of oxidizing trace organics in reclaimed wastewater (Gerrity et al., 2008).
Recovery and release of TiO2 across the ceramic membrane
Potential passage of TiO2 across the ceramic membrane into the treated water was investigated. Figure 9 shows the concentration of TiO2 in the ceramic membrane permeate for 0.1 g/L TiO2 in 5 mM NaHCO3 deionized water as well as in dechlorinated tap water; all samples were taken at a run time of 15 min and analyzed by spICP-MS. The permeate concentrations depended on water matrix and illumination (3–4 lamps operating). Without illumination, TiO2 was poorly recovered by the ceramic membrane. Increasing irradiation significantly improved TiO2 recovery, indicating photoaggregation of the TiO2 as larger aggregated particles were captured by the ceramic membrane and less permeation occurred. There are only a few reports indicating that illumination of catalysts results in aggregation (Liu et al., 2014; Sun et al., 2014), what we term here as photoaggregation, but results indicate TiO2 surface hydroxyl groups enhance aggregation, which results in changes to photocatalytic properties and longevity of the catalyst.
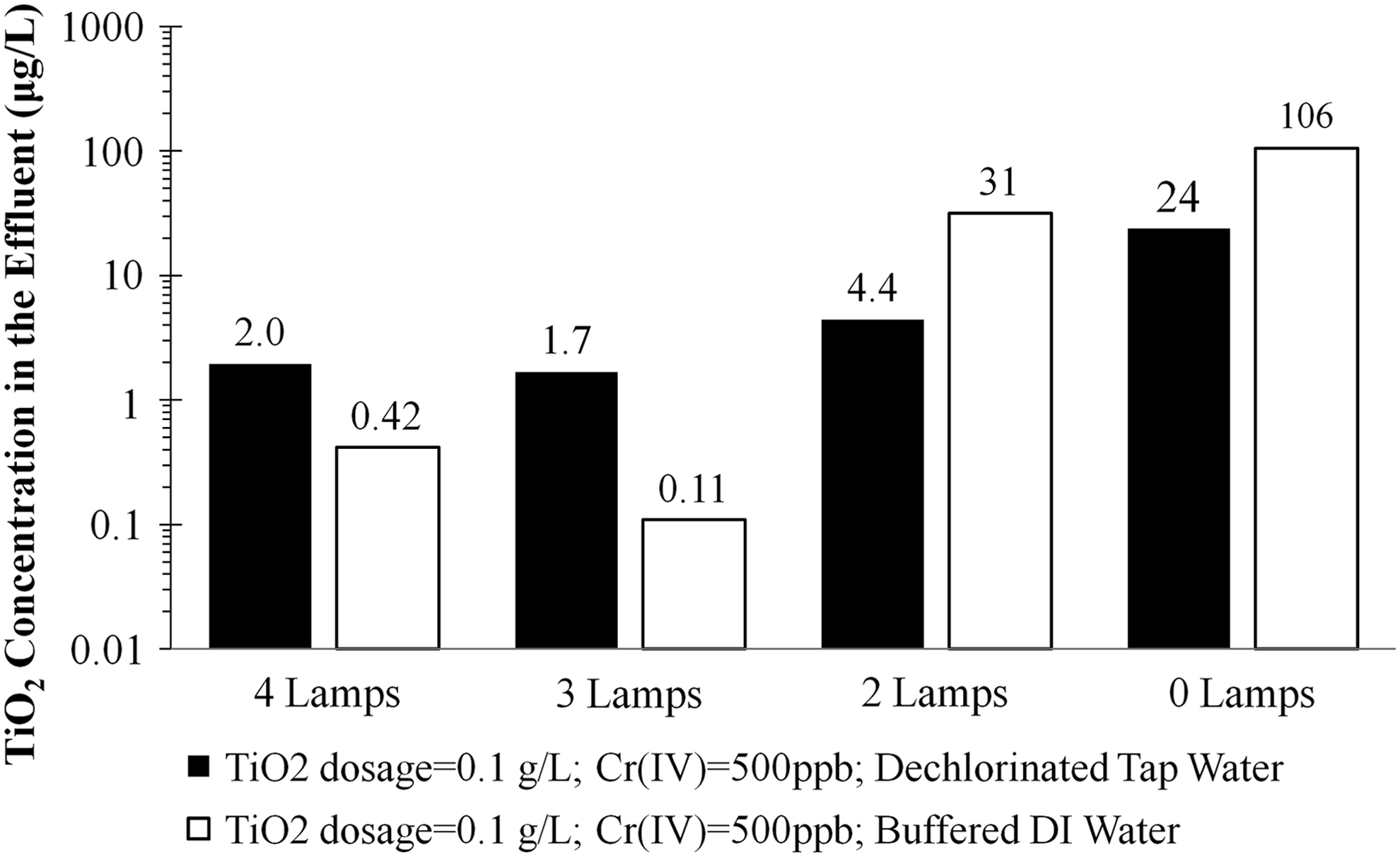
Titanium dioxide concentration in membrane permeate samples taken at t=15 min as a function of lamps and water matrix. pH for 5 mM NaHCO3-buffered DI matrix increased from 8.5 to 8.7 from Cin to Cf; pH for dechlorinated tap water increased from 7.7 to 7.9 from Cin to Cf.
Without illumination of tap water, 24% of the TiO2 passed the ceramic membrane. With any level of illumination in tap water, less than 5% of the TiO2 passed the membrane. Tap water contains roughly 5 μg/L of Ti, but the spICP-MS mode clearly indicated TiO2 particles in the membrane permeate that were not present in the initial tap water, that is, above the background concentration. Primary particle size of well-dispersed P90 TiO2 is 12–18 nm. Because pores in ultrafiltration membranes range from 0.001 to 0.1 μm (1–100 nm), some pores may be larger than the individual TiO2 particles, leading to particle breakthrough.
Reasons for observed variations in leaching may be TiO2 aggregation due to UV illumination, which induces diminished electrostatic forces between particles and generates bridging hydroxyls on the surface (Sun et al., 2014); pH and ionic strength differences of the water matrix, which can impact aggregate morphology and fractal dimension (Domingos et al., 2009; Chowdhury et al., 2013; Tong et al., 2013); ionic aqueous constituents such as Ca2+, which promote aggregation (Chowdhury et al., 2012; Liu et al., 2013); or decreased ability to pass the ultrafiltration membrane upon higher chromium surface loading onto TiO2 and thus increased size of TiO2 particles. Significant catalyst aggregation diminishes the photoactive surface area of the particle and increases likelihood of charge carrier recombination before surface reactions, which subsequently weakens the ability for photoinduced reduction of hexavalent chromium, a concern for scalability for long irradiation durations and large water volumes.
Conclusions
Removal of hexavalent chromium was investigated for several water matrices, titanium dioxide dosages, and energy inputs using an engineering-scale photocatalytic reactor system. The integrated UV-TiO2/ceramic membrane system reduces Cr(VI) and removes all aqueous chromium species. Catalyst dosage was the most impactful parameter investigated, with the most successful dosage being 1 g TiO2/L. Higher alkalinity, pH, or presence of divalent salts decreased the effectiveness of Cr(VI) adsorption and subsequent reduction, which required higher EE/O. The presence of divalent cations in the tap water likely aggregated the TiO2. P90 has primary particle sizes of 12–18 nm, which are of the same size or smaller than some ultrafiltration membrane pores.
Agglomeration of the initial media or aggregation of TiO2 during use increases its size, making passage through the membrane less favorable. This phenomenon may occur as the catalyst produces electrons and holes on the TiO2 surface during illumination, thus changing the local surface charges and allowing aggregation to occur. Additional research is needed to understand this photoaggregation process alone as well as on the role of divalent salts and the role of TiO2 photoaggregation on this advanced photo-oxidation process.
Surface analysis confirmed that chromium species were present on the surface of the TiO2 in the effluent slurry and significant aggregation of TiO2 particles when in flow-through mode. As Cr or inorganic foulants accumulate on TiO2, catalyst effectiveness is reduced as evidenced in the flow-through experiments. Based on full-scale implementation data, recovery and regeneration of the spent TiO2 may be completed through acid washing the media to remove Cr(III) species and foulants, allowing for reuse of the TiO2. While Cr(VI) photocatalytic reduction occurs readily in ultrapure water at low irradiance levels, the presence of salts, alkalinity, and elevated pH increases energy requirements. These factors must be overcome for full-scale implementation, and feasibility studies must determine regeneration rates for the catalyst to maintain optimal running conditions. Overall, photocatalytic reduction of Cr(VI) appears feasible in complex water matrices after managing aggregation and foulants. Combined reduction and removal through photocatalytic processes for drinking water treatment may alleviate chromium exposures and risk of adverse health effects while addressing new regulatory limits.
Footnotes
Acknowledgments
This research was supported by the National Science Foundation (CBET 1132779). Graduate student support was partially provided by a Dean's Fellowship from the Ira A. Fulton Schools of Engineering at Arizona State University. Materials were characterized in the LeRoy Eyring Center for Solid State Science at Arizona State University.
Author Disclosure Statement
No competing financial interests exist.