
Abstract
Abstract
Long-term atrazine degradation was studied with Pseudomonas sp. strain ADP (P. ADP) encapsulated in electrospun microtubes with no addition of external carbon source. Experiments were conducted in consecutive 3-day sequential batches under semisterile and nongrowth conditions over a period of 2.5 years. During the entire period, 95.2% atrazine was degraded on average, with 68.2% nitrogen recovery as ammonium. Only a slight buildup of intermediate (approximately 15–20% cyanuric acid) was observed. Confocal microscopy taken after 6 months showed bacteria colonizing outside the microtubes whose origin came from external contamination. Analysis of the microtube microbial community after 1.5 years revealed a mixed population containing P. ADP along with Chitinophagaceae, Variovorax, and Microbacterium. After 2.5 years, P. ADP was not found in the microtube bacterial population comprising Variovorax, Cupriavidus, Comamonas, Xanthobacter, Microbacterium, and Tsukamurella. Presence of pADP-1 plasmid in the microtube microbial population after 2.5 years indicates horizontal plasmid transfer from the initial P. ADP population to other bacteria. This suggests that gene transfer from a single bacterium, P. ADP, capable of using atrazine as a nitrogen source, but not as the energy source, allowed for the evolution of a mixed culture capable of degrading atrazine and using it both as a nitrogen and energy source. Results from this study show potential benefits of using microtube encapsulation of bacteria for bioremediation of recalcitrant compounds.
Introduction
A
In spite of its persistence in soil and water supplies, atrazine can be broken down in the environment by microorganisms. Several bacteria have been shown to carry atzABCDEF genes and trzNDF homologs on one or more self-transmittable plasmids, with dehalogenation being the first stage of the metabolism (Smith et al., 2005). Bacterial consortia, with members separately holding the relevant genes, have also been shown to catabolize atrazine.
The best-characterized atrazine-metabolizing bacterium studied is Pseudomonas sp. ADP (P. ADP) isolated from a herbicide spill site (Mandelbaum et al., 1995). While atrazine's side chains of ethylamine and isopropyl amine can be used as an electron donor, literature reports that P. ADP can use atrazine only as a nitrogen source but not as a carbon source (Topp et al., 2000; Neumann et al., 2004). P. ADP has been shown to rapidly mineralize atrazine completely under aerobic, denitrifying, and nongrowth conditions and shows the potential for use in bioremediation schemes (Mandelbaum et al., 1995; Katz et al., 2001; Wyss et al., 2006). Enriched cultures of P. ADP have been successfully used in biofilm reactors for atrazine treatment of groundwater under aerobic conditions using citrate as the carbon source.
Cell encapsulation technologies may provide a platform for atrazine's bioremediation needs without the need for adding a carbon source. Encapsulation of microbial cells promises to enhance the stability of bacteria, protect bacteria from being washed out and from mechanical or chemical damage. In addition, bacterial encapsulation can sustain a large population for extended periods, while providing the ease of separation from the operational environment. Most microbial encapsulation methods presently available have used polymeric materials (polyacrylamide, polysaccharide, and hydrogels), ordered mesoporous silica, or sol–gel systems. In general, these methods have shown important shortcomings, such as low viability of the microbes, insufficient diffusion through the material, nonuniform cell distribution, poor thermal and mechanical stability, and loss of biological activity (Zussman, 2011).
Electrospun microfibers are currently under investigation as a potential alternative cell encapsulation technique. Characteristics, such as high porosity, microscale interstitial space, and large surface-to-volume ratio, make electrospun microfibers attractive for biomedical applications, such as tissue engineering, carriers for biologically active molecules, including proteins and enzymes, and environmental engineering applications, such as membranes for particle removal (Xie and Hsieh, 2003; Patel et al., 2006; Ramakrishna et al., 2006). However, encapsulation of biologically active materials, such as proteins, enzymes, and cells in electrospun fiber, can be problematic. The characteristic features of the process, such as the extremely rapid formation of the microfiber, as well as the high elongation rate (resulting in cross-sectional area reduction), create very large shear and tensional stresses on biologically active materials. In addition, the presence of toxic solvents in the polymer solutions can potentially harm the bioactive agent.
Core–shell microtube technology may offer a suitable solution since in the single-step coelectrospinning process, the potentially toxic organic phase, containing a water-insoluble polymer (the outer shell solution), is separated from the aqueous phase (the core solution) (Dror et al., 2007). To date, this cospun microtube technology has been applied in laboratory scale for pure enzyme encapsulation (Dror et al., 2008) and bacterial cell encapsulation for atrazine degradation (Klein et al., 2012). However, it was shown that the encapsulation process had a negative effect on bacterial activity, and the microtube shell fabricated with polycaprolactone (PCL) was susceptible to biodegradation (Klein et al., 2012).
This article examines long-term durability and atrazine degradation of P. ADP bacterium encapsulated in core–shell electrospun microtubes in consecutive batch experiments under semisterile conditions without the addition of a carbon source. These operating conditions have the potential advantages of minimizing water contamination and eliminating intensive posttreatment typically required in groundwater bioremediation processes.
Materials and Methods
Chemicals
Atrazine (2-chloro-4-ethylamino-6-isopropylamino-s-triazine), 94% technical grade was acquired from Agan Chemicals, Ashdod, Israel. Hydroxyatrazine (97% purity) was purchased from Riedel-de Haën, and high-performance liquid chromatography (HPLC) grade cyanuric acid was purchased from Sigma-Aldrich Chemicals.
Encapsulation of P. ADP cells in electrospun microtubes
Encapsulation of bacteria in electrospun microtubes was carried out according to Klein et al. (2012) with poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF–HFP) as the shell solution instead of PCL to increase durability and polyvinylpyrrolidone (PVP) as the core solution. P. ADP bacteria were grown and enriched according to Mandelbaum et al. (1995). P. ADP bacteria were cultured in a growth solution, harvested by centrifugation, and washed three times with autoclaved double-distilled water. The final pellet was resuspended in 0.5 mL distilled water and added to the PVP core solution, 1:3 (vol/vol). To increase cell viability, freshly spun microtubes with P. ADP were collected from the surface of an autoclaved phosphate buffer bath (PBS ×1) by winding on a partially immersed plastic carrier rotating in the bath.
Atrazine degradation assay
To examine the long-term activity of microtubes with encapsulated P. ADP under nongrowth conditions, consecutive sequential batch experiments were conducted over a period of 2.5 years. Four carriers with microtube coverings were placed in duplicate 100-mL Erlenmeyer flasks on an orbital shaker at 25°C filled with 50 mL of 20 mM phosphate buffer containing ∼20 mg/L atrazine (BPA solution). After each batch, the carriers were washed twice with 20 mM phosphate buffer and once with a BPA solution. Batches were changed twice a week with a fresh BPA solution. Semisterile conditions were maintained during the long-term sequential batches, that is, all solutions were sterilized before use, and the filling and washing of Erlenmeyer flasks were carried out on a microbiological bench with flame to avoid contamination. Initial and final concentrations of atrazine were measured by spectrophotometer (García-González et al., 2003), and the amount of atrazine removed was calculated. At the end of the batch, the ammonium concentration was measured as mg/L N by colorimetry (Willis et al., 1996). The percent N recovered was calculated by dividing the ammonium concentration as N found at the end of the batch by the stoichiometric amount of nitrogen in the initial atrazine concentration (32.5% of atrazine concentration or ∼6.6 mg/L as N for an initial atrazine concentration of 20.2 mg/L). The chloride concentration was determined by ion chromatography (761 Metrohm ion chromatograph equipped with 150-mm Metrosep A Supp 5 column and precolumn; Metrohm AG, Herisau, Switzerland) using an eluent containing 3.2 mM Na2CO3 and 1.0 mM NaHCO3.
Atrazine and its metabolites were detected by HPLC equipped with a diode array detector (Agilent Varian ProStar). Fifty-microliter samples were injected into a Phenomenex C18 column (100 A 150 × 4.6 mm, particle size 5 μm) and monitored at 220 nm. The mobile phase was kept constant at 5% acetonitrile for 7 min, followed by a linear gradient to 60% acetonitrile for 20 min.
Scanning electron microscopy
Images of the fibers were obtained using an FEI E-SEM Quanta 200 scanning electron microscope (SEM).
Characterization of immobilized P. ADP
Immobilized bacteria were characterized by epifluorescence microscopy using a Zeiss Ikon Axioskop-40 equipped with a 63×/1.25 oil objective lens (EC plan-Neofluar) and CCD camera (AxioCam ICc 3). Live/Dead BacLight Bacterial Viability Kit (Invitrogen) and BacLight RedoxSensor CTC Vitality Kit (Invitrogen) were used according to the user's manual instructions.
Flow cell experiments
The flow cell system consisted of a feed flask (500-mL Duran bottle), tubing, a peristaltic pump, a bubble trap, a flow chamber (model number 81; BioSurface Technologies Corporation), and a waste flask (500-mL Duran bottle). The required equipment was sterilized with 70% alcohol for 30 min and then autoclaved for 30 min. One hundred microliters of three times washed enriched culture from microtubes was used as inoculum and injected into the bubble trap through a rubber membrane. The bacteria were allowed to settle in the flow chamber for sufficient adhesion for 15 min. Sterile phosphate buffer with atrazine (BPA medium) was then pumped to the flow chamber at 0.7 mL/min. The flow chamber was mounted on the stage of optical microscope (Olympus IX50 with an LD 60× phase contrast PH2 objective) equipped with a CCD camera. Images were captured at a rate of 12 frames per hour and stored in hard disk.
Microbial community analysis with polymerase chain reaction–DGGE and sequencing
Total genomic DNA of the original P. ADP culture and microtube cultures (encapsulated bacteria) after 1.5 and 2.5 years were extracted using FastDNA SPIN Kit for Soil (MP Biomedicals) following the manufacturer's protocol. A 0.5-mL pellet collected from the P. ADP culture and ∼0.5-g wet microtube material were used as samples. The DNA concentrations of the extracts were measured with the NanoDrop 1000 Spectrophotometer (Thermo Scientific), adjusted for polymerase chain reaction (PCR) amplification, and stored at −20°C until further use.
PCR was performed in a thermocycler (TProfessional Basic Gradient Thermocycler; Biometra). Apex RED Taq Master Mix (Genesee Scientific Corp.) was used to amplify the variable region V3–V5 of the bacterial 16S rDNA using the primer pair consisting of 341F (5′-CCTACGGGAGGC AGCAG-3′) with a GC clamp (5′-CGCCCGCCGCGCCCCG CGCCCGTCCCGCCGCCCCCGCCCG-3′) and 907R (5′-CCGTCAATTCCTTTRAGTTT-3′) (Muyzer et al., 1993). The PCR conditions were as follows: initial denaturation at 95°C for 5 min, 28 cycles with primer annealing at 58°C for 1 min, DNA elongation at 72°C for 1 min, denaturation at 95°C for 45 s, followed by final primer extension of 10 min at 68°C. PCR products were evaluated for purity by horizontal agarose electrophoresis on a 1.5% (wt/vol) agarose gel.
Denaturing gradient gel electrophoresis (DGGE) was performed using the DCode Universal Mutation Detection System (Bio-Rad Laboratories), as previously described (Muyzer et al., 1993). Equal volumes of PCR product (30 μL/well) were loaded on a 6% wt/vol polyacrylamide gel in TAE buffer. The DGGE was performed at 60°C using a denaturing gradient ranging from 35% to 70% and a constant voltage of 100 V. After a 17-h run, the gel was stained for 30 min with 0.4 μg/mL ethidium bromide (Amresco LLC) and washed for 15 min with distilled water. DNA bands were visualized and photographed with an UV illuminator (UVP LLC). The dominant DGGE bands were carefully excised with a sterile scalpel (Albion) under UV illumination and placed in 20–30 μL PCR reagent water for DNA extraction (Sigma-Aldrich). After overnight incubation at 4°C, the gel was pelleted by centrifugation at 13,000 g for 5 min. For DNA reamplification, 1 μL of the supernatant was used as a template for PCR. Conditions and reagents of the run were as initially described above using primer set (341F and 907R) without a GC clamp. The sequencing of PCR products was carried out by Hy-Labs, Israel. The resultant sequences were submitted to the NCBI GenBank (www.ncbi.nlm.nih.gov/blast) database for identification.
Plasmid extraction and agarose gel electrophoresis
A piece of microtube was inoculated in an atrazine growth medium containing citrate as a carbon source. The bacteria were collected after 3 days by centrifugation at 14,000 rpm for 15 min. A QIAGEN Miniprep Kit was used to extract plasmid DNA from the resulting pellet along with pADP-1 plasmid extracted from a pure P. ADP culture. 0.7% agarose gel was prepared in 2× TAE buffer. Ten microliter sample volume with 3 μL loading dye were loaded on the gel, and electrophoresis was carried out at 60 mA and 120 V for 2 h or until the dye neared the end of the gel. The gel was placed in 0.4 μg/mL ethidium bromide (Amresco LLC), and the DNA bands were visualized and photographed with an UV illuminator (UVP LLC) (Meyers et al., 1976).
Results
Long-term performance of microtubes encapsulated with P. ADP in consecutive batch experiments for atrazine degradation under nongrowth semisterile conditions
Consecutive batch experiments were conducted on microtube-encapsulated P. ADP with no carbon source addition. In all, 248 batches were conducted over a period of 2.5 years (3 to 4 days per batch). Atrazine degradation was high, about 95% (Table 1). During the experimental period, a nitrogen recovery (measured as ammonium) of 68% of the theoretical amount available from atrazine was observed, indicating the presence of atrazine metabolites at the end of the batch.
A representative batch of 4 days without carbon addition after 1.5 years of consecutive work is shown in Fig. 1 and shows atrazine degradation and ammonium and atrazine metabolites' formation. During the initial 10 h of the batch, 96.5% of the atrazine was removed and completely biodegraded in less than 30 h. Eighty-five percentage of the theoretical amount of chloride available from atrazine was measured in 10 h. The hydroxyatrazine and cyanuric acid concentrations rose to 5 and 3.6 mg/L, respectively, in 10 h. After 10 h, hydroxyatrazine concentration slowly decreased, reaching zero after 56 h with 100% chloride recovery. A residual concentration of 2.2 mg/L cyanuric acid was observed at the end of the batch, and 77% of ammonium was recovered. Increasing the length of the batch did not increase cyanuric acid degradation. The nitrogen in the remaining cyanuric acid and hydroxyatrazine accounted for 1.1 and 0.4 mg/L as N, respectively, and nearly balanced the theoretical nitrogen yield together with the ammonium produced. No adsorption of atrazine or by-products on the surface of microtubes was found.

Atrazine degradation showing metabolites during a representative batch experiment.
Long-term changes in microtube appearance
After the first 6 months of consecutive batches of atrazine degradation, the microtubes were examined with confocal microscopy. CTC stain (red) was used to show actively respiring cells, while SYTO 9 (green) was used to enhance the appearance of the microtube walls and stained all cells (Boulos et al., 1999). Areas of dense microbial population inside the microtubes were observed, suggesting growth of bacteria, while other areas had moderate or no bacterial population (Fig. 2A, B). In addition, the outer surface of the microtubes also showed a strong fluorescence, suggesting that growth had occurred and a biofilm had developed (Fig. 2B).

Epifluorescence micrographs
After 2.5 years of consecutive batches, the surface morphology of the microtubes was examined using SEM. The microtubes appear intact, demonstrating their long-term durability, while the pores in the shell of microtube were still visible and not clogged (Fig. 2C). In spite of the nongrowth conditions in the sequential batch experiments, a well-developed biofilm was evident on the outer surface of microtubes (Fig. 2D).
Flow cell experiments
After the observed appearance of an external biofilm using confocal microscopy, flow cell experiments were conducted to record any bacterial growth, even though no external carbon source was added. The flow cell was inoculated with bacteria from the microtubes and continuously fed with BPA for 3 days. Division was observed after a lag phase of 37 h and may be due to the long time it took the bacteria to degrade atrazine and use their intermediate products for growth. After the lag phase, division occurred every 25 min. The first two generations are shown during the 37th hour in Fig. 3A–M, and a colony appeared after 3 days' time (Fig. 3N).

Series of selected micrographs (magnification 600×) showing microtube culture dividing in flow cell during exponential phase. More than 800 such images were generated in an experiment [hour:min from start of batch:
Microbial community analysis after 1.5 and 2.5 years
DNA was first extracted from two sources: (1) pure culture of P. ADP grown in enriched medium (atrazine medium) and (2) encapsulated bacteria used for more than 1.5 years in the consecutive batch experiments under semisterile conditions, with atrazine as the only substrate and no addition of carbon source. DNA extracts were subjected to the PCR–DGGE analyses. The DGGE profile of the enriched pure P. ADP bacteria showed one band, whereas microtube DGGE consisted of four dominate bands (bands 1–4) (Fig. 4A). The enriched culture was identified as genus Pseudomonas (586 bp), 100% identical to strain ADP (DSM 11735). Individual bands on the DGGE of the microtube bacterial population were identified by their closest similarity with 16S rDNA sequences stored in NCBI GenBank and belonged to the genera Chitinophagaceae, Variovorax, Pseudomonas, and Microbacterium. Except for Chitinophagaceae, the remaining bacteria have been reported to degrade atrazine (Table 2). After 2.5 years of consecutive batches, the microbial community analysis of the microtubes showed further diversity (Fig. 4B and Table 3); however, the original inoculant P. ADP was not found.
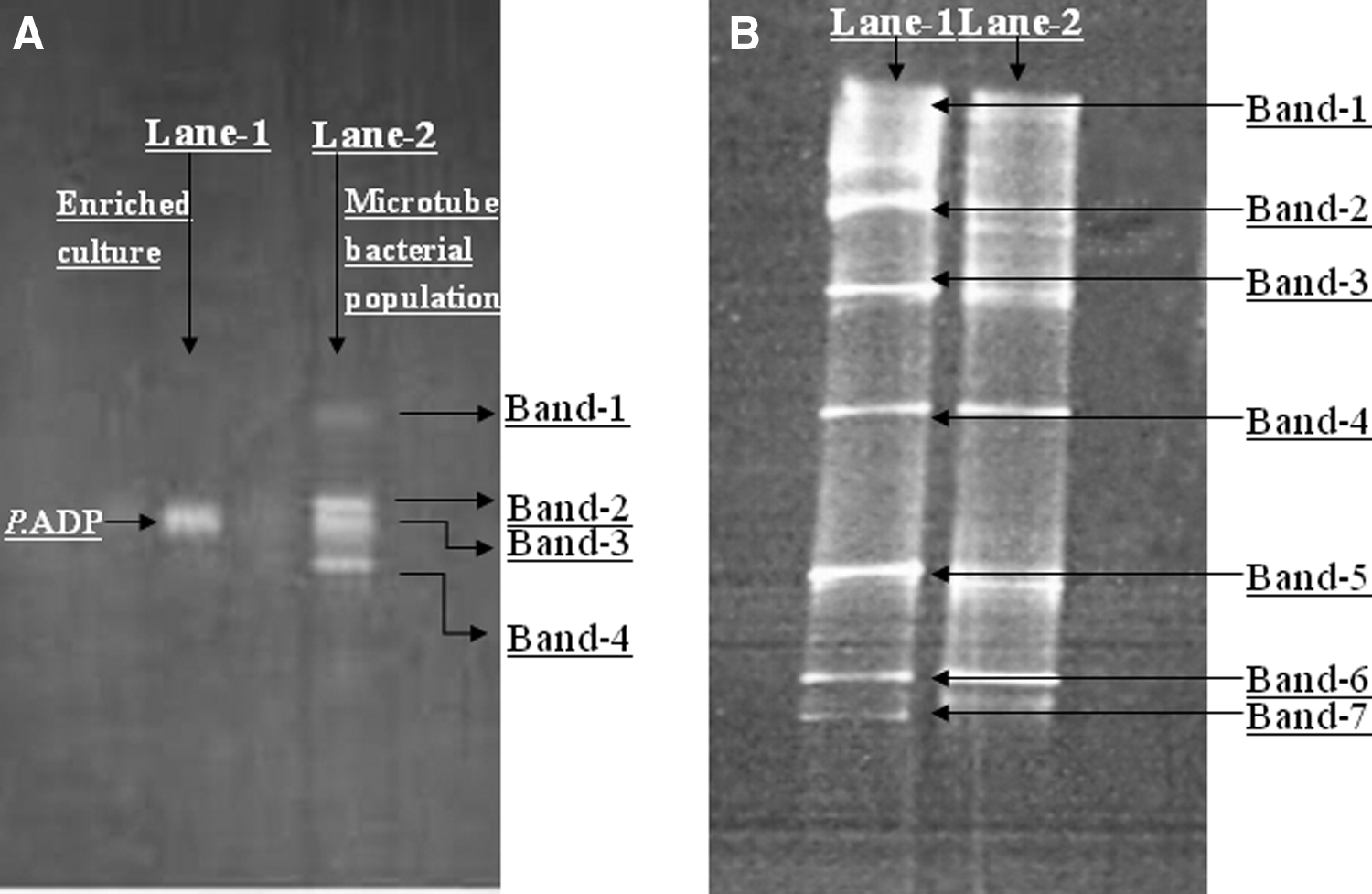
DGGE, denaturing gradient gel electrophoresis.
Plasmid profile
Absence of P. ADP from the microbial community of the microtubes prompted the examination of the presence of the pADP-1 plasmid responsible for atrazine degradation in P. ADP. Plasmid DNA was extracted by growing a small piece of microtubes on atrazine medium with citrate and compared with pADP-1 plasmid from pure P. ADP bacterium using agarose electrophoresis (Meyers et al., 1976) (Fig. 5). Agarose gel electrophoresis confirmed the presence of pADP-1 plasmid in the 2.5-year-old microtube culture.
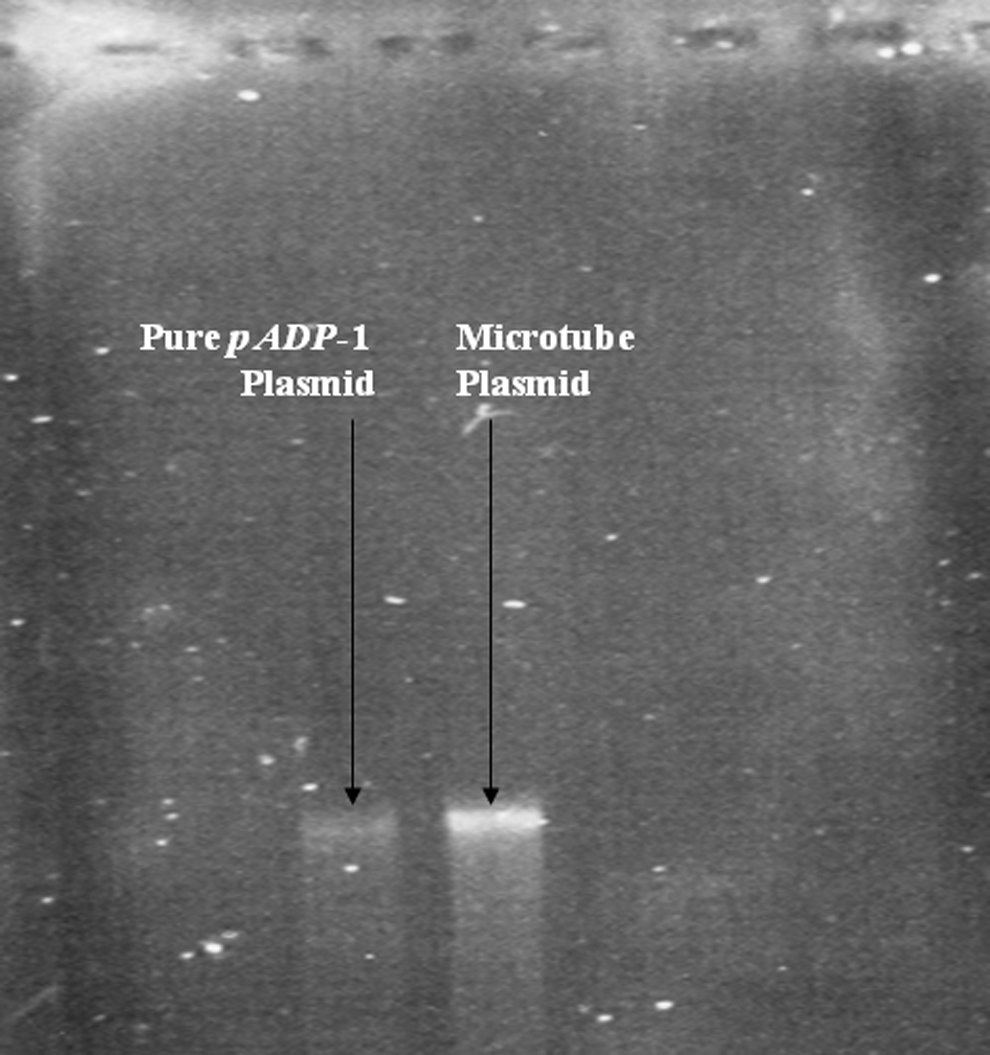
Agarose gel electrophoresis of plasmid DNA; left side: pure pADP-1 plasmid; right side: plasmid from microtubes.
Discussion
Results of nearly 250 sequential batches over a 2.5-year period clearly demonstrated that core–shell electrospinning encapsulation of P. ADP resulted in an active bacterial population with a long-term ability to degrade atrazine alone without any external carbon source addition. The degradation of atrazine was shown to be nearly complete (95.2% ± 6.8%), with only minimal residual concentrations of atrazine, hydroxyatrazine, and cyanuric acid remaining at the end of each batch. Similar results were observed with several other shorter term sequential batch experiments starting with P. ADP encapsulated in electrospun microtubes (results not shown).
The change in shell formulation from PCL to PVDF–HFP prevented potential microtube deterioration (Klein et al., 2012) and resulted in long-term durability. In addition, the collection of freshly spun microtubes in a buffered water bath significantly increased cell viability, and an initial period in a growth solution to recover atrazine-degrading activity was not necessary as before (Klein et al., 2012). However, over time, confocal and SEM microscopy confirmed the existence of bacteria colonizing outside the microtubes whose origin came from either microtube leakage or external contamination.
Sustained removal of atrazine during the sequential batch experiments without loss of degradation ability was particularly notable because P. ADP is not known to use atrazine as a carbon source (Topp et al., 2000; Neumann et al., 2004). In addition, the other potential carbon sources tested here (the core solution of the microtubes and dead cells from the electrospinning process) were found not to support growth. While atrazine does contain usable carbon in its side chains, the solubility of atrazine significantly limits side-chain concentration for bacterial growth, and only a few bacteria have been found that use atrazine as a sole carbon source (Yanze-Kontchou and Gschwind, 1994; Topp et al., 2000). Flow cell experiments inoculated with bacteria from the microtubes confirmed microbial growth on atrazine only, suggesting bacterial contamination other than P. ADP.
Comparison of the DGGE from the initial pure culture of P. ADP and after 1.5 years showed a significant change in the bacterial population from one band to four, proving that P. ADP was not the sole bacteria present. At the end of the experimental period (2.5 years), P. ADP was not found in the bacterial population, clearly demonstrating that another bacterium or a bacterial consortium was responsible for atrazine degradation in the batch experiments. Only one isolate (Comamonas sp. A2) sequenced was known to be able to degrade atrazine completely as P. ADP and uses it similarly as a nitrogen source only but within the confines of a bacterial consortium (Yang et al., 2010).
Nearly all the isolates found have been previously recovered from bacterial consortium degrading atrazine, supporting the idea of cooperative degradation of the herbicide (Tables 2 and 3). However, the presence of a single plasmid similar in size to pADP-1 plasmid and the disappearance of P. ADP from the microtube population at the end of the experimental period strongly suggest plasmid transfer from the initial P. ADP population. Therefore, complete atrazine degradation could be carried out by single microorganisms rather than in the case of a consortium, where genes responsible for atrazine degradation are distributed among various organisms. This is supported by Devers et al. (2007), who demonstrated the transfer of a genetically modified version of pADP-1 plasmid to Variovorax sp. in soils.
s-Triazine herbicides, such as atrazine, were introduced to the environment during the 1950s. Initially, these compounds were known to be poorly biodegraded with long half-lives of several hundred days (Shapir et al., 2007; Krutz et al., 2010). The intensive use of these xenobiotic recalcitrant compounds led to the recent evolution of microbial populations capable of rapid mineralization of these compounds in different parts of the world, either by single mineralizing bacterium of diverse bacterial genera or by microbial consortia. In comparison, the changes in the microbial population observed in the consecutive batch experiments reported here over a much shorter period were unexpected, particularly under oligotrophic conditions due to the low solubility of atrazine.
Summarizing, initial atrazine degradation in the extended consecutive batches was probably carried out by the large population of resting P. ADP cells (Mandelbaum et al., 1995) freshly encapsulated in microtubes. The release of ethylamine and isopropylamine side chains from atrazine degradation to solution and not metabolized by P. ADP provided enough energy for microbial contaminates to slowly form a biofilm and acquire atrazine-degrading capability by pADP-1 plasmid transfer. Bacterial contamination may have occurred when the microtubes were originally collected in an exposed water bath during electrospinning (although the buffer solution was sterilized) or during the many batch washings and fillings that required opening and closing of the flasks.
The above observations may have significant implications in the design of bioremediation techniques for atrazine and atrazine-like compounds. For example, water systems typically contain low bacterial concentrations, and therefore, similar results would be expected when using encapsulated bacteria, such as P. ADP, for atrazine bioremediation in potable water supplies. Microtubes or other encapsulation techniques can provide the physical housing and protection during the initial inoculation of large numbers of bacteria with genes capable of biodegrading recalcitrant compounds. Over time, bacterial contaminants originating from the water source with a competitive advantage can proliferate and form biofilm on the backbone of the encapsulating material and through gene transfer from the original inoculant, gain the ability to biodegrade a given compound, such as atrazine. As a result, a much more stable and robust microbial community can develop over time.
Conclusions
In this research, electrospun core–shell microtubes offered a stable and durable platform for the encapsulation of the atrazine degrader P. ADP. Nearly complete atrazine degradation was observed for 248 consecutive batches over a 2.5-year period. However, under conditions of atrazine only, where P. ADP is not known to grow, a major shift in microbial population was observed, and a biofilm developed on the microtubes. The bacterial community that developed was shown to grow on atrazine alone and contained the P. ADP plasmid, strongly suggesting plasmid transfer as the mechanism for long-term sustained atrazine degradation. Microbial contamination and biofilm formation may be difficult to prevent in electrospun microtube applications; however, microtubes can provide the structure for the attachment and development of a rich and stable microbial population, where preferred genetic information can be transferred and disseminated from the original encapsulated bacteria.
Footnotes
Acknowledgments
This study was supported by the German–Israeli BMBF/MOST Water Technology Research Fund, grant no. WT1103/GR2336, and the China–Israel Cooperative Scientific Research Program.
Author Disclosure Statement
No competing financial interests exist.