
Abstract
Abstract
We have used solid-state nuclear magnetic resonance (NMR) of 29Si to monitor the reaction of forsterite (Mg2SiO4) with isotopically enriched 13CO2. Fate of silicon that comes from dissolution of forsterite in the presence of water or NaCl brine was monitored as a function of depth in a packed bed reactor, which used elevated-temperature and -pressure conditions to mimic underground geological sequestration conditions. Silicon provides an important window into the reaction since the precipitation of amorphous silica can affect forsterite dissolution, and solid-state magic-angle spinning NMR can readily distinguish between 29Si in forsterite and amorphous silica. Furthermore, differences between Q3 and Q4 species of amorphous silica were observed as a function of depth. Results differed between water- and brine-based reactions, with enhanced dissolution of forsterite observed in the brine.
Introduction
F
Lackner et al. (1995) were perhaps the first to suggest forsterite carbonation for CO2 mineralization. Hanchen et al. (2006) demonstrated that forsterite dissolution is the first rate-limiting step, and magnesite (MgCO3) precipitation is the second rate-limiting step in the overall process of carbonation of olivine. There are multiple factors that can affect the dissolution processes, such as temperature, pH, and CO2 concentration (Pokrovsky and Schott, 2000; Oelkers, 2001; Daval et al., 2011). Critically, the dissolution of the mineral is inhibited by a passivating silica layer, whose structure and morphology are not well characterized, that formed on the surface of forsterite (Daval et al., 2011) and of olivine (Sissmann et al., 2014) during the reaction with CO2.
Our goal has been to characterize the chemical species (carbon based and silicon based) present during a reaction of CO2 with a forsterite (Mg2SiO4) slurry in a packed bed reactor, in the presence of water or NaCl-brine. Nuclear magnetic resonance (NMR) is an inherently element-selective, nondestructive form of spectroscopy. NMR is a powerful characterization tool, in that it monitors the local bonding environment surrounding an isotope of interest, and, consequently, does not require crystalline regularity for detection.
In the past, we conducted in situ NMR in specialized hardware that we had constructed (Surface et al., 2013) that allows for the simultaneous detection of solid, liquid, gaseous, and supercritical phases of 13C-bearing species in a packed bed reactor. Here, we are reporting on NMR conducted “ex situ,” performed on solids collected from the in situ NMR probe, using the high resolution afforded by the technique of “magic-angle spinning” (MAS) NMR (Andrew, 1971) to average out interactions in the solid state that broaden the resonances. In doing so, we can then have the combination of both measurement schemes—comparing results from in situ studies with higher-resolution spectra from ex situ MAS NMR to refine the results from static experiments.
We have recently reported on the formation of both aqueous bicarbonate and solid magnesium carbonates in situ (Moore et al., 2015), with simultaneous detection of solution-phase and solid-state 13C NMR resonances. We have also used NMR as a part of a comprehensive investigation of carbonate mineral formation in packed beds of forsterite, complemented by Raman spectroscopy and reactive transport modeling (Giammar et al., 2014).
Consequently, the objective of this complementary study is to evaluate the 29Si solid-state NMR of forsterite samples that were reacted at elevated temperatures and pressures (to mimic GCS conditions) in both water and a brine of NaCl in water. Similar to diffusive transport in tubular reactor experiments that we have previously reported (Giammar et al., 2014; Xiong and Giammar, 2014), here, we have reacted a packed bed of forsterite with CO2-water or CO2-brine that can be analyzed as a function of depth in the sample. We have been able to detect (semi-quantitatively) with 29Si NMR both the silicate mineral dissolution and the precipitation of amorphous silica as a function of depth in the reacting column.
Experimental Protocols
Forsterite (Mg2SiO4) powder (Alfa Aesar; Stock No. 43807), sample size of 1.902 g, was mixed with 1.138 mL ultrapure water (resistivity 18.2 MΩ-cm) to make a slurry and loaded into the NMR sample space (a cylindrical space that was 4.3 cm in length and 10 mm in diameter) as previously reported (Surface et al., 2015). In reactions with NaCl, a 1.0 M solution (in ultrapure water) was mixed into the forsterite. The sample, a slurry, was pressurized and maintained with a pressure transducer at 100 atm 13CO2 (99% 13C-labeled gas; Sigma-Aldrich), and the temperature was set to 100°C. After equilibrating at that temperature and pressure for several hours, the reaction was allowed to proceed for 25 days. (The sample that was reacted with the 1.0 M NaCl solution was reacted for 29 days.). The solids remaining after the reaction were removed from the liquid, dried, and apportioned into 2 mm-thick disks for (ex situ) solid-state NMR analysis of the 29Si species. For example, the sample labeled “2 mm” extends from the top of the sample (0 mm) to 2 mm below the surface.
Figure 1 is a schematic of the sample space in the homebuilt elevated-temperature and -pressure in situ NMR probe, which is able to perform with reaction conditions (temperature up to 250°C, pressure up to 300 bar) that are meant to mimic geological sequestration environments. As shown in the schematic, the packed bed of forsterite was separated into 2 mm-thick samples, and those analyzed here were at the following depths (from the top of the reaction bed): 0–2, 4–6, and 14–16 mm.
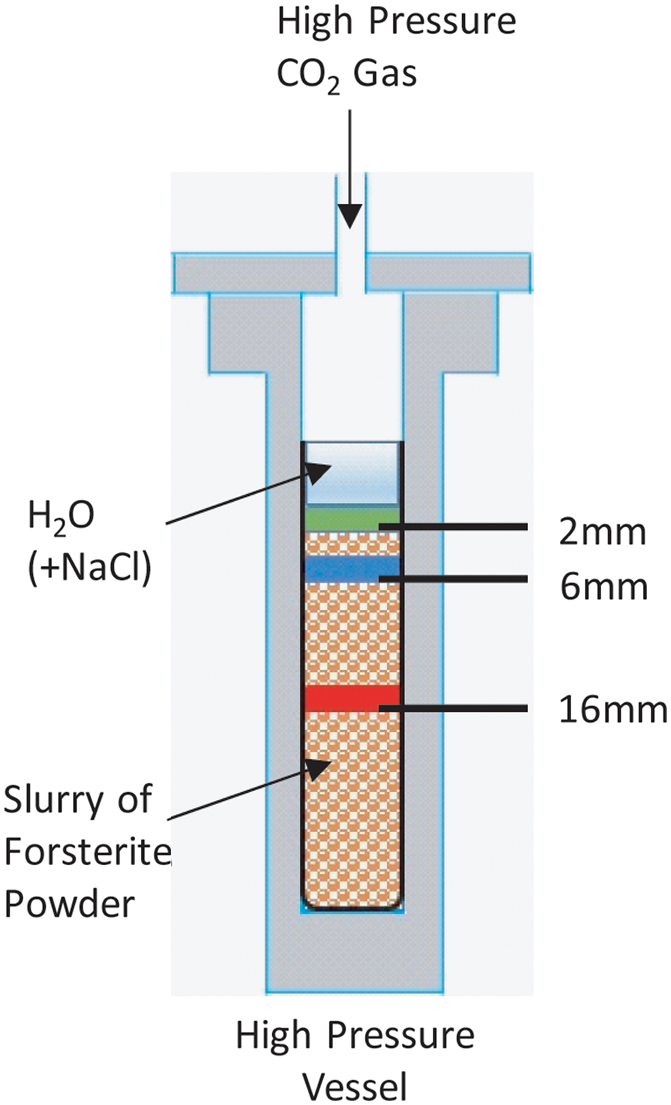
Schematic of in situ reaction vessel for forsterite slurry and elevated-pressure 13CO2.
29Si {1H} MAS NMR
Solid-state 29Si{1H} MAS NMR (with proton decoupling) was used to probe the silicon-containing reaction products from the dissolution of forsterite (The use of proton decoupling was necessary to observe resonances from the amorphous silica that were broadened by nearby protons.). The experiments were performed in a commercial HXY Chemagnetics MAS 4 mm probe on a Tecmag spectrometer at a resonance frequency of 58.6 MHz for 29Si (with 294.97 MHz for 1H decoupling). MAS was carried out at a 14 kHz rotation frequency. Typical data acquisition used four-step phase cycle spin-echo experiments with π/2 pulse lengths of 4.4 μs, a τ delay of 136.2 μs, and recycle delays of 390 s, which is approximately twice as long as the T1 time for forsterite and, by all indications, is in excess of 5 × T1 for amorphous silica (detailed in the next paragraph). The number of transients recorded was 800 per 29Si{1H} spectrum. 1H decoupling was used in the experiments with B1 field strengths of 18 kHz; in the absence of decoupling, portions of the amorphous silica spectra were obscured by heteronuclear dipolar coupling to nearby protons. The spectra were referenced to tetramethylsilane (TMS) at a 29Si chemical shift of 0.0 ppm.
NMR data integration was completed using the peak fitting software DMfit. All the peaks were fit using Gaussian functions.
T1 spin-lattice relaxation time was measured for four of the six samples reported here. Details can be found in the Supplementary Data (Supplementary Figure S1a–c). The forsterite T1 for the three samples with the highest signal-to-noise ratios are 136–153 s; a fourth sample with a much weaker signal has such a poor signal-to-noise ratio as to undermine the accuracy of the measurement (all data can be seen in the Supplementary Data). All the amorphous silica have shorter T1 times, which are measured as 19–80 s.
Results
Figure 2 is a diagram of sites typically found in silica and silicates, which are categorized by the bonding environment of the silicon and labeled by the terms “Q1, Q2, Q3 and Q4.” A Q4 silicon has four Si-O-Si bonds without any protons; a Q3 site has three Si-O-Si bonds and a single Si-OH bond; a Q2 site has two Si-O-Si bonds and two Si-OH bonds; and a Q1 site has just one Si-O-Si bond and three Si-OH bonds. Forsterite has been previously studied by 29Si NMR (Ashbrook et al., 2007; Stebbins et al., 2009; Kwak et al., 2010), and the resonance observed at approximately −61 ppm is representative of SiO44–tetrahedra that are interspersed with the Mg2+ cations, sometimes referred to as “Q0” units, following the nomenclature described earlier. It has been well established that the Q1 to Q4 sites in amorphous silica may be assigned based on their isotropic chemical shifts, falling into ranges that reflect the presence of protons as well as the O-Si-O bonding angles (Magi et al., 1984).

Schematic for nomenclature used for sites present in silica: Q1, Q2, Q3, and Q4.
Figure 3a shows the 29Si {1H} MAS NMR spectra for the reaction of forsterite with 13C-labeled 13CO2 in the absence of NaCl. The resonance at −61.70 ppm was assigned to forsterite, similar to prior reports (Kwak et al., 2010). It has a narrow linewidth (0.74 ppm) that agrees well with these reports of 29Si NMR of forsterite, consistent with a polycrystalline high-purity solid. A broad resonance centered at approximately −111 ppm is typical for Q4 of amorphous silica. A Q3 resonance at around −102 ppm was also observed (notably only in the presence of proton decoupling, not observed with 29Si MAS). Figure 3b shows the 29Si {1H} MAS NMR spectra for the same reaction mixture in the presence of NaCl. The smaller peak areas for the forsterite mean more extensive dissolution near the top of the packed bed. These peak areas were recorded with recycle delays that are approximately two to three times the T1 relaxation time (390 s recycle delay versus the measured T1 at 135–153 s) because of the need to record many transients and the inability to signal average over very long periods (A recycle delay of 765 s would be required for the spectrum to be quantitative.). So the peak areas are not quantitative, but they may be used qualitatively.
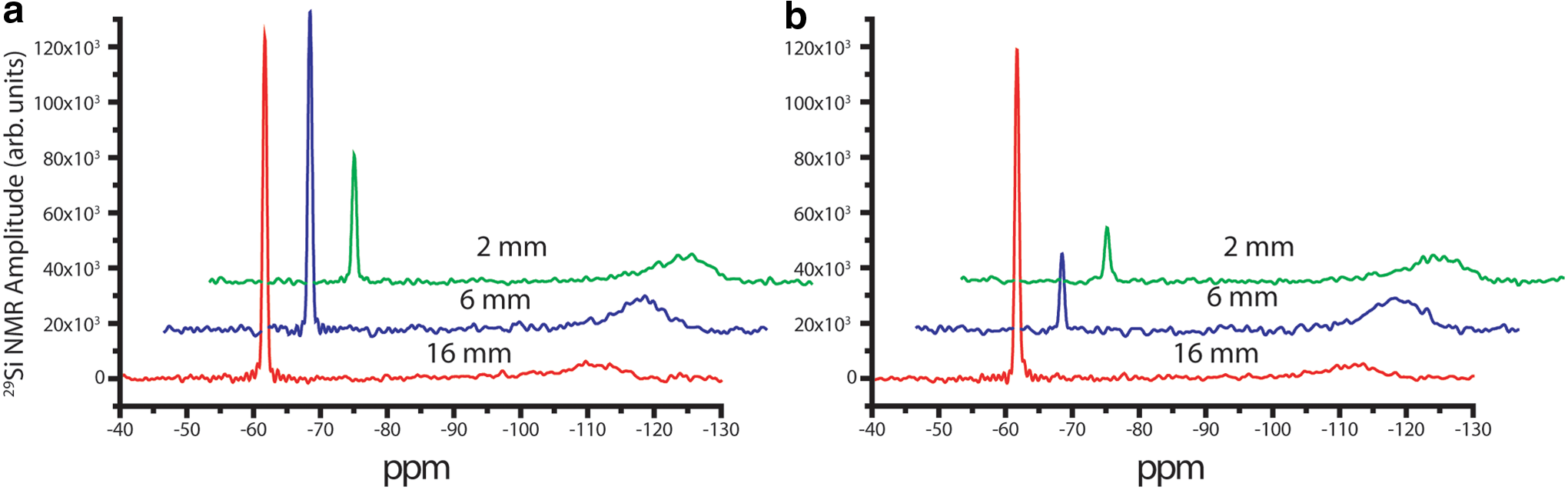
29Si{1H} MAS spectra with 1H decoupling for reacted forsterite at sample depths of 2, 6, and 16 mm
Though not rigorously quantitative—which would require a recycle delay of 5 × T1 between transients—the relative areas of these peaks can be readily compared (The relative intensities within the series shown in Fig. 3a and b are meaningful, as well as the comparison between them.). Table 1 gives values for the integrated area of the (reactant) Mg2SiO4 29Si resonance and the precipitated amorphous 29Si resonances for each of the slices (The amorphous silica region was deconvoluted into two Gaussians—shown later in Fig. 4—and the peak area reported here is the sum total of the fits.). The amorphous silica that is formed (as a percentage of the total 29Si signal) through dissolution-precipitation is larger higher up in the column, as shown in Table 1, which matches the trend found for the forsterite dissolution, with more dissolution found closer to the surface of the packed bed.
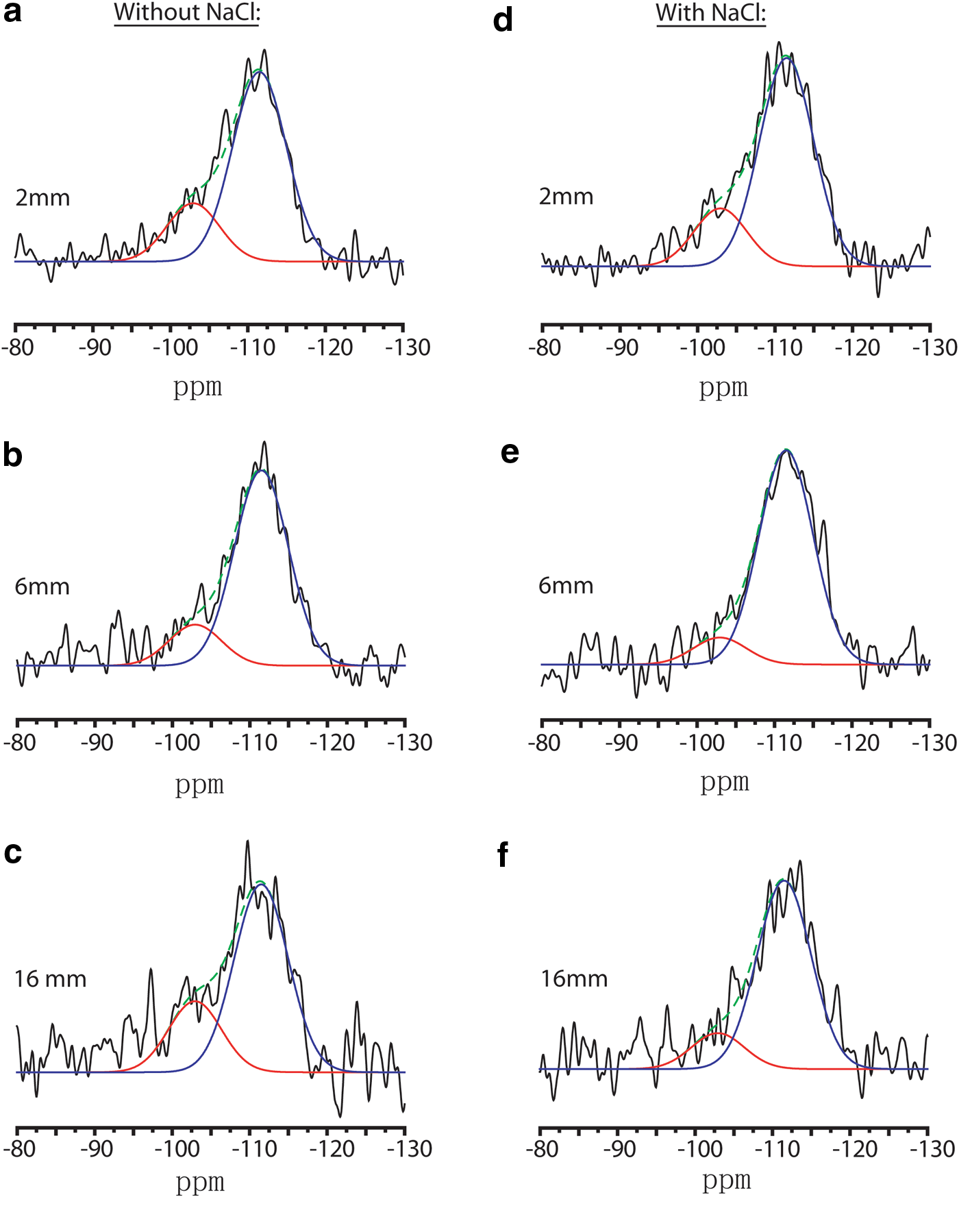
Deconvolution of amorphous silica 29Si{1H} MAS nuclear magnetic resonance (NMR) resonances by DMfit into two Gaussian lineshapes.
Forsterite mineral “starting material” columns give ranges of areas, taking into account that they were recorded with recycle delays less than 5 × T1. Areas have been scaled from ∼2 × T1 to 5 × T1 based on their projected magnetization buildup.
The extent of reaction in both water and brine varies with a function of depth in the packed bed. The NMR evidence for this is given by the integrated peak areas for the 29Si{1H} solid-state NMR.
Different amorphous silica products were observed both with and without NaCl. The water-based slurry favored formation of both Q3 and Q4 silica, whereas the brine-based slurry favored Q4 silica sites (overall) and led to enhanced dissolution of the forsterite for similar reaction times and identical conditions. Figure 4 shows the deconvolution of the amorphous silica 29Si NMR resonance into multiple Gaussian peaks. With the help of {1H} decoupling, we could identify Q3 sites. However, Q1 and Q2 resonances are not evident here. 29Si{1H} CPMAS spectra were recorded and are shown in the Supplementary Data (Supplementary Fig. S2). From those spectra, we know of the presence of Q3, but we use 29Si{1H} MAS NMR when hoping to quantify species. To best resolve the carbonation reaction in brine versus a water-only slurry, we use the DMfit program to deconvolute the amorphous silica resonances into Q3 and Q4 sites.
A Q3 species was fit to both sets of samples, whereas the water-only packed bed had more intense Q3 resonances overall (seen readily “by eye” in Fig. 4). In the NaCl-brine, Q3 sites were fit to the amorphous region based on the same chemical shift and peak width; however, there is only minor evidence of its presence given the low signal-to-noise ratios for Q3 in the 6 and 16 mm samples.
Discussion
29Si solid-state NMR has a set of experimental challenges that makes this isotope less commonly studied. Both mineral species such as forsterite (Mg2SiO4) and Q4 sites in amorphous silica suffer from a lack of protons that would be exploited for cross-polarization experiments. Consequently, 29Si experiments are complicated by samples with long T1 spin-lattice relaxation times, making quantitative characterization very difficult. Here, we have conducted a series of experiments with a relatively long recycle delay (390 s) to achieve sufficient signal-to-noise ratios to qualitatively probe the identity of silicon-containing reaction products from forsterite dissolution, including some (i.e., amorphous silica) that have been challenging to identify using other techniques. Our NMR data show that we are observing forsterite dissolution based on the decrease in the peak area and the concomitant increase in the amorphous silica region as one product of the reaction.
Mineralization for systems with pH < 8 is based on dissolution-precipitation reactions and can be explained by the following coupled chemical equations:
The formation of the condensed amorphous silica layer can be envisioned as the condensation/polymerization and dehydration of the orthosilicic acid, H4SiO4. The resulting silica is polymerized on the surface of forsterite when the Mg is dissolved and removed from the mineral, yielding the amorphous layer. This phenomenon has been observed for forsterite dissolution at conditions with pH <9 (Pokrovsky and Schott, 2000). In 2011, Daval et al. observed the formation of amorphous silica layers of 15–40 nm by transmission election microscopy (TEM), which could cause the dissolution rate of olivine to dramatically decrease. In experiments with forsterite dissolution in the presence of high-pressure CO2, declines in dissolution rates beyond what could be explained by the changes in surface area and pH with reaction extent were attributed to the likely formation of such a layer (Wang and Giammar, 2013). The amorphous silica on the surface is believed to stop the formation of MgCO3 (Bearat et al., 2006), which is unfavorable in conditions where mineralization is desirable.
Figure 3a and b shows a diminution of forsterite dissolution as a function of depth in the column of the reacting slurry—as evidenced by the intensity of its 29Si NMR resonance. The dissolution of forsterite was expected to be the most extensive in the regions closest to the CO2-saturated water, because the pH will be the lowest in this zone. More orthosilicic acid is released from the packed bed in the 2 and 6 mm samples. These 29Si data also follow the approximate trend of MgCO3 formation (evidenced by 13C NMR Supplementary Fig. S3; both data sets are shown in Supplementary Fig. S4), with the maximum carbonate formation observed between 4 and 8 mm, lessening deeper into the packed bed (We must state this with a caveat, because we did not ensure that the 13C NMR data are rigorously quantitative owing to the extremely long T1 times for magnesium carbonate, by some estimates as much as 4 h.). If T1 is unchanging throughout this series, the data (taken with a single transient after magnetizing for 45 min per experiment) are consistent for the series, and the peak intensities are representative of the quantity of carbonate present. This trend is notable, because it follows predictions from the Xiong and Giammar (2014) diffusion-limited transport model, which predicts a specific spatial distribution of carbonate.
Data in Fig. 3 demonstrate the dramatic differences observed in the presence of NaCl: The NaCl leads to a greater extent of forsterite dissolution but not to a correspondingly greater amount of amorphous silica precipitation. A larger amount of forsterite dissolved in the 2 and 6 mm samples in brine compared with the reaction in water, indicating that NaCl facilitates the dissolution of forsterite. These observations are consistent with an earlier batch dissolution study of forsterite in CO2-saturated solutions that found greater extents of dissolution in NaCl solution than in a more dilute solution (Wang and Giammar, 2013). We surmise that in the process of mineralization, the pH drops on CO2 exposure, and the Na+ present reacts with Si-OH to form Si-ONa (Simakin et al., 2008), which could inhibit the formation of amorphous silica by preventing its condensation polymerization from forming the silica network. As a result, more orthosilicic acid is released from the packed bed in the 2 and 6 mm samples. Notably, the 6 mm sample in Fig. 3b does show a somewhat larger29Si amorphous silica resonance compared with others in this packed bed, but it is not substantially larger with respect to the water-only sample. These results suggest that although more forsterite has reacted at 6 mm in the presence of NaCl brine, it does not lead to a concurrently larger amorphous product.
There is increasing pH as a function of depth, creating conditions that differ at the surface. For the samples in the packed bed, the pH will increase with depth, favoring deprotonation of Q3,–SiO–over–SiOH.
Although there was significant forsterite dissolution in the water-based slurry, in the topmost layer the amorphous silica was not proportionally greater. In fact, the amorphous silica component is similar between 2 and 6 mm samples, even though there is significantly less dissolution deeper in the packed bed. It is possible that at the top (0–2 mm), the orthosilicic acid (H4SiO4) is able to diffuse out of the sample. A similar process of reactant diffusion out of the topmost layers of the packed bed has been suggested by reactive transport modeling (Giammar et al., 2014) to limit magnesite precipitation in this layer, with the greatest magnesite accumulations occurring somewhat deeper into the bed.
In the NaCl-brine, the 2 and 6 mm samples have very similar amounts of forsterite dissolution and amorphous silica present. The distinguishing feature between these two is that Q3 species are not observed to an appreciable extent deeper in the packed bed. Interestingly, the surface 0–2 mm samples are relatively similar between water-only and NaCl-containing solutions. Deeper in the column at 16 mm for both samples, the smallest amount of amorphous silica is observed, which is due to the higher pH and lower dissolution rate of forsterite.
Q3 and Q4 sites are present at different ratios as a function of depth. Previous studies (Dove and Elston, 1992; Icenhower and Dove, 2000) have shown that the dissolution of amorphous silica was affected by the pH, metal cation(s) present, and temperature.
At the surface of the packed bed, the pH of the solution is estimated to be ∼3.2 (Xiong and Giammar, 2014), which will affect the topmost 2 mm sample. Consequently, the Q3 silica will be the most stable (Dove and Elston, 1992; Berger et al., 1994). Therefore, these conditions lead to the highest relative Q3 content for both samples.
Summaries
We have undertaken an NMR study of two packed beds of forsterite that were reacted with 13CO2: one with a water slurry and one with a 1.0 M NaCl brine slurry; reacted with elevated-temperature and -pressure conditions. We have evaluated 29Si solid-state NMR as a function of depth for three samples from the packed bed: 0–2, 4–6, and 14–16 mm.
Fate of the silicon from forsterite (Mg2SiO4) is important since the precipitation of amorphous silica can affect forsterite dissolution. Here, we have monitored the 29Si NMR to show both dissolution of the forsterite and precipitation of amorphous silica—as both Q3 and Q4 silica species. NaCl-containing brine enhances dissolution of forsterite deeper in a packed bed of the mineral than water alone. The water-only reactions favor formation of both Q3 and Q4 amorphous silica, whereas there is less Q3 silica in the presence of the brine. NaCl appears to inhibit the formation of the Q3 species. Even with the enhanced dissolution of forsterite in NaCl-brine, it is notable that the amounts of amorphous silica found were not concomitantly larger.
We have demonstrated that solid-state NMR is a valuable analytical tool for monitoring the disposition of silica in mineralization reactions, including amorphous species that are not readily probed by other analytical techniques. There is potential for in situ 29Si NMR experiments, even with the experimental complications detailed earlier. The temporal evolution of forsterite dissolution could be monitored, given its narrow linewidth, and dissolved silica species could be monitored in the solution phase. The effect of brine solutions on mineralization can be determined through such NMR experiments, ultimately leading to the evaluation of conditions for better CO2 sequestration reactions in heterogeneous systems such as these.
Footnotes
Acknowledgments
This material is based on work supported by the Department of Energy under Award No. DE-FE0023382. This article was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied; assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed; or represents that its use would not infringe privately owned rights. Reference here to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of the authors expressed here do not necessarily state or reflect those of the United States Government or any agency thereof. The design of initial experiments with NaCl solutions benefited from discussions with Fei Wang. Early experiments were conducted with funding from Washington University's Consortium for Clean Coal Utilization.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.