
Abstract
Abstract
Injection and subsequent carbonation of calcium and magnesium silicate particles are proposed here as a novel strategy to grout fractures or pores and decrease fluid conductivity in the deep subsurface by taking advantage of favorable reaction kinetics. This approach could be used to manage legacy environmental risks from energy extraction techniques such as hydraulic fracturing of shale formations. To evaluate this approach, we studied the carbonation of wollastonite (CaSiO3) in the presence of ground shale under temperature and pressure (T/P) conditions broadly representative of the gas-producing regions of the Marcellus formation in the eastern United States. Effect of calcite (CaCO3) precipitation on shale particle morphology and interparticle pore structures was investigated and impacts on permeability were evaluated using batch and column experiments. Formation T/P were positively correlated with extent of carbonation. Side reactions involving calcium ions and passivation by amorphous silica did not appreciably impede the carbonation reaction. Quantitative X-ray diffraction results showed conversions in excess of 50% after 24 h, indicating that many shale plays possess the necessary T/P conditions for favorable carbonation kinetics. Scanning electron microscope–energy dispersive X-ray results showed that CaCO3 precipitates effectively cemented unconsolidated shale particles. Mercury intrusion results indicated that the porosity of column reactors packed with shale particles decreased appreciably after reaction with CaSiO3 and CO2-saturated water. These CO2-mediated dissolution/precipitation reactions could form the basis of permeability control technologies to manage emerging risks of fluid leakage and contaminant migration from energy production and waste disposal activities in the deep subsurface.
Introduction
S
The boom in shale gas extraction has been enabled largely by two technologies, horizontal drilling and hydraulic fracturing (Kerr, 2010). The hydrocarbon-bearing zones in shale formations typically exist several kilometers beneath the surface in relatively thin strata (on the scale of 10s of meters), so horizontal drilling technologies have been pioneered that allow operators to contact a large areal extent of those zones from a single well pad. Pressurized aqueous fluids are then forced through perforations within these horizontal well segments to create dense fracture networks that cut across gas-conducting bedding planes. Proppants, most often sand, are used to keep the fractures open during fracture fluid flow back and hydrocarbon production stages (Weaver et al., 2005). After production, these flow paths could enable fluid migration and contaminant transport into overlying sedimentary formations where faults and abandoned wells could then conduct these fluids into near-surface formations, posing a long-term risk to groundwater resources (Darrah et al., 2014).
Use of the term “shale” in the context of unconventional oil and gas production refers to low permeability sedimentary rocks with highly variable mineral composition of compacted clays, quartz, feldspar, and carbonates (Chermak and Schreiber, 2014). Organic fraction characteristics vary as a function of age and distribution within the formation, but the total organic content of most hydrocarbon-bearing shales is generally less than 10% by mass. Shale also contains toxic metals and radionuclides and high concentrations of arsenic, barium, and radium in drill cuttings and flow back wastewater (Barbot et al., 2013; Wang et al., 2015). Fluids move through artificial and natural fractures with apertures on the order of millimeters to micrometers (Palmer et al., 2013). Because of the depositional nature of shale formations, fractures often grow longer laterally rather than vertically (Fisher et al., 2012). The bulk shale formation has pores with diameters on the order of 10s of nanometers (10−9 m) (Ross and Marc Bustin, 2009). These nanopores are important in terms of the excess sorption capacity of the bulk shale rock but less critical from a standpoint of decreasing fluid connectivity within a fractured shale network. Although intact shale has fluid permeability on the order of nD (10−9 Darcy) (Heller et al., 2014), fractured shale can have an effective permeability on the order of 10–100 mD (10−3 Darcy) (Cho et al., 2013; Kumar et al., 2016).
Efforts to control fugitive emissions of residual methane or seepage of fluids from depleted shale formations would need to limit fluid flow through these fractures once the wells have been abandoned. This study provides a proof-of-concept for a method that could dissolve proppants made of mineral silicates and subsequently fill these fractures by precipitating minerals that reduce formation permeability. Such a method could address environmental challenges associated with preventing contaminant migration from the formation once hydrocarbon extraction is complete and a site is closed. The mineral carbonation reactions employed in this method have the secondary benefit of storing injected CO2 in the formation, thereby mitigating some portion of the climate implications of shale gas development (Edwards et al., 2015). In particular, we set out to determine whether Ca-bearing silicate minerals, used as proppants or injected once hydrocarbons have been extracted, could be reacted with injected CO2 after production but before a well is retired and closed to seal the fracture network with carbonate minerals. A schematic of the concept is provided in Fig. 1.
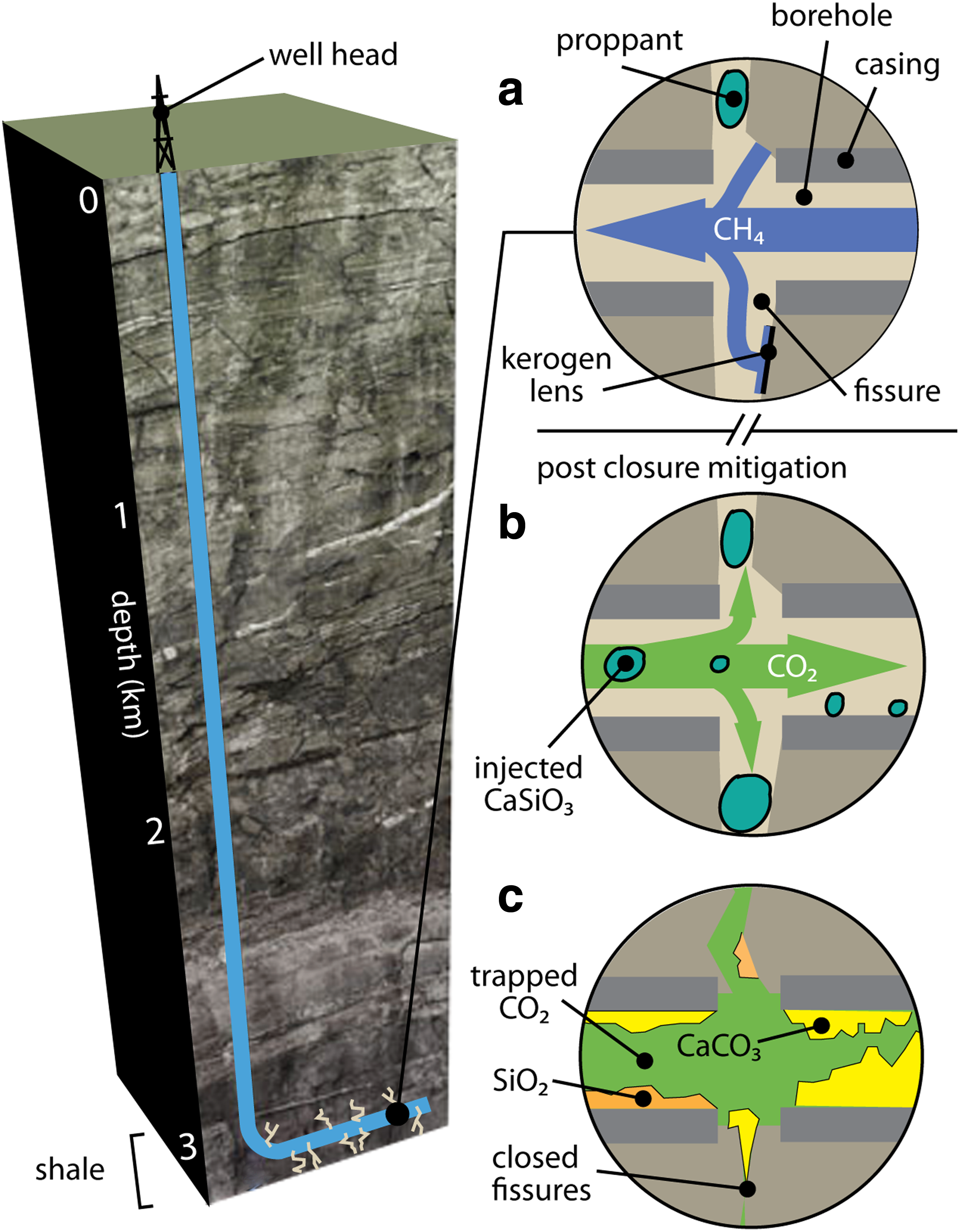
Schematic illustrating how mineral carbonation reactions would be used to mitigate many of the risks that could result from shale-fracturing activity. Fracture networks and proppants that are initially put in place to produce natural gas
Ex situ mineral carbonation reactions have been studied over the past decades as a method to mineralize CO2 captured from power plants (Gerdemann et al., 2007). Magnesium- and calcium-containing silicate minerals such as olivine (MgSiO3) and wollastonite (CaSiO3) react readily with CO2 under high temperature and pressure (T/P) conditions to form solid magnesium or calcium carbonate and silica through the following reaction (Xiong and Giammar, 2014):
where
These reactions are spontaneous at STP but the kinetics are slow and so most experimental work has reported on conversion at T/P conditions in the range of 100–175°C and 15–25 MPa (Huijgen et al., 2006; Gerdemann et al., 2007). Much of the literature on ex situ deployment of mineral carbonation have focused on reactor design and conditions for optimal conversion. A mechanistic understanding of the chemistry of these reactions was proposed by Huijgen et al. (2006), who proposed a passivation model for relating the particle size of the cation donor with the fluid phase properties. This model suggests that the dissolution of the silicate would be the rate-limiting step but that the crystal structure of carbonate and silica precipitates could affect the reaction rate significantly (Daval et al., 2009). The resulting competition between dissolution and precipitation processes is both temperature and pressure dependent such that optimal conditions exist for driving the reaction toward completion (Huijgen et al., 2006). Given the emphasis on optimizing batch reaction conditions, little effort has been applied to understanding how these reactions would proceed in the environment under more complex chemical or mass transfer-limited conditions. In contrast, if silicate carbonation reactions could be carried out in situ within deep subsurface formations, many of the technical and economic obstacles associated with ex situ deployment of this approach could be resolved. In particular, shale formations exist at depths where the geothermal gradient and the lithostatic pressure typically approach optimal T/P needed to achieve mineral carbonation reactions. In addition, reaction rate kinetics would be important but not as important as they are in ex situ conditions where reactor residence time is limited.
A major limitation to the ex situ carbonation approach is that the cost and environmental burdens associated with extracting the mineral feedstock, dissolving the cation donor mineral, and reacting the flue gas at high T/P are likely to outpace the benefits in emissions reductions (Khoo et al., 2011). Some studies have explored the possibility of carrying out the reaction in the subsurface (i.e., in situ carbonation) in regions where these minerals predominate. Work by Matter and Kelemen (2009) and Giammar et al. (2014) suggests that CO2 injection into formations with high concentration of olivine or calcium/magnesium-bearing silicate minerals (such as basalt) would be an effective method for mineral sequestration of CO2 at sites with inadequate structural trapping. The primary challenge in developing injection strategies with mineral trapping as the primary goal is to ensure that fluid conductivity is preserved during the CO2 injection phase. Several pilot projects have been deployed to advance this concept, including the Carbfix program in Iceland and the Big Sky Regional Partnership in the United States, which includes injection into basalt formations (Gislason et al., 2010; McGrail et al., 2012; Gislason and Oelkers, 2014). Preliminary results from these trials suggest that the carbon storage potential of these formations is promising.
The idea of using depleted shale formations to permanently sequester CO2 has been proposed in recent years (Tao and Clarens, 2013). Most efforts to develop geologic CO2 sequestration have focused on deep saline aquifers because of their large estimated capacities and broad geographic distribution, although sequestration in shales has a number of important benefits (Busch et al., 2008). Notably, hydrocarbon extraction creates significant volumes of pore space, much of which preferentially sorbs CO2 over CH4 (Tao and Clarens, 2013; Edwards et al., 2015). Most shale gas extraction operations would also already be connected to gas transport systems (e.g., pipelines) capable of delivering captured CO2 at relatively low cost (Middleton and Bielicki, 2009). Furthermore, the potential for sequestering CO2 by forming stable mineral precipitates would potentially improve the safety and viability of CO2 sequestration in shales (Matter and Kelemen, 2009).
Stoichiometrically, mineral silicates are consumed on a 1:1 basis with CO2, which means that the mineral demands for sequestration using mineral carbonation requirements would be large. In practice, a significant fraction of CO2 would be sorbed to kerogen surfaces, filling many of the surface binding sites and nanopores that would be otherwise filled with CH4. Moreover, a significant amount of free dense-phase CO2 could still exist in the formation and be trapped (as illustrated in Fig. 1) by fractures and pores grouted with CaCO3 and SiO2. This conceptual model of trapping requires that pockets of CO2-enriched brine do not dissolve the mineral precipitates. The geochemistry would be complex, but recent modeling studies focused on understanding dissolution of wellbore cements and carbonate-rich rocks in the presence of CO2-rich brines suggest that such trapping processes are possible under certain conditions (Raoof et al., 2012; Nogues et al., 2013).
Other strategies for reducing permeability in the context of CO2 sequestration or hydraulic fracturing operations have been proposed. The injection of water into overlaying aquifers or beneath caprocks has been suggested as one method to retard leakage of buoyant gases in the context of CO2 sequestration (Zahasky and Benson, 2016). Another approach is the use of pH-sensitive silica gel, which can decrease the permeability of the leaking fractures and block flow paths containing high partial pressures of CO2 (Druhan et al., 2014, 2015). One area of study that has received considerable attention is leakage pathways through cement materials because of the importance for understanding gas seeps in the annular region of the wellbore. Cao et al. (2015) observed the changes in aperture of a fracture in a cement sample caused by a CO2–brine mixture and reported a decrease in permeability because of the incidence of carbonation reactions. Luquot et al. (2013) also reported the decrease of fracture width under low flow rate of CO2-rich brine, suggesting that cement has self-healing properties. This work revealed an important influence of flow rate and its effect on aperture, suggesting that complex dynamics exist between the chemistry and the physics of flow in these systems. Tongwa et al. (2013) evaluated the feasibility of using paraffin wax, polymer-based gel, silica-based gel, and microcement as engineered strategies to seal fractures in the context of CO2 sequestration and reported that each of these is suitable under different conditions. Both gel and wax are less resistant to pressure after sealing when compared with microcement. Overall, this body of work suggests that strategies to cement fractures would provide one of the most stable methods to trap fluids in formations, like shales, that are not naturally abundant in available divalent cations.
To assess our overarching goal of developing a permeability control method using mineral carbonation reactions under the in situ conditions characteristic of the deep subsurface, we sought to test five hypothesis: (1) mineral carbonation reactions would proceed spontaneously under the T/P conditions of a representative shale formation, (2) mineral and organic components of the shale will not lead to side reactions that significantly inhibit carbonation reactions, (3) the kinetics of the reactions are relatively rapid and would proceed to completion in days to weeks, (4) the precipitation reactions will take place in spatial proximity to the dissolution reactions, and (5) the precipitates will decrease the porosity and permeability of a porous medium that is otherwise unreactive with aqueous CO2 in the absence of a divalent cation donor mineral. In this article, we report on both batch and flow through experiments that were used to test these five hypotheses. To ensure the reproducibility of our results, our experiments were carried out using a model benchtop system involving packed columns filled with ground and well-sorted shale powders. This setup allowed us to recreate the physicochemical conditions of fractured shale formations to explore the relationships between chemical reactions and flow under idealized conditions.
Experimental Protocols
To evaluate the behavior of silicate dissolution/precipitation reactions within a porous shale, we tested the physicochemical characteristics of these reactions within stirred batches and columns of ground size-sorted shale at high T/P conditions. This experimental configuration was not intended to duplicate the physics of a deep subsurface shale formation, in which natural or artificial fractures provide flow paths within the bulk formation matrix. Instead, it was designed to provide reproducible experimental conditions that would enable us to create combinations of capillary numbers and reactant particle sizes that are comparable with what would be observed within a fracture network in the subsurface. This setup provided two advantages over carrying out the experiments in fractured core samples. First, mineralogical heterogeneities within the rock could be evenly distributed throughout the samples to minimize experimental artifacts associated with differences in the rock composition. Second, the effects of precipitation reactions on porosity within the shale particle packed columns could be quantified and more easily extrapolated to a large spatial extent than would be possible using a fractured shale core. Using ground shale particles as a surrogate for a fractured rock did introduce some characteristics to the experimental setup that would not be expected in the field. Most notably, the tortuosity and roughness of the flow paths would likely be lower in a fracture network. To minimize the effect this would have on our experiments, the particle size of the ground samples was selected such that the characteristic length scales of the pores were representative of fracture networks in the field.
Ground shale particles used in the experiments had diameters on the order of 10 μm and pore spaces on the order of 1–10 μm (Bertei et al., 2013). Recent work has used artificial fractures in rocks to characterize the role of dissolution/precipitation reactions (Ellis et al., 2011; Deng et al., 2013; Giammar et al., 2014). But for reasons related to reproducibility and chemical heterogeneity already listed, we chose to explore the physicochemical characteristics of these reactions in packed columns of granular shale samples with a focus on exploring the possibility of deploying carbonation reactions within the pore spaces between shale particles (i.e., interparticle), not the nanopores present within the shale grains (i.e., intraparticle). When selecting grain sizes for the experiments here, we took care to create conditions in which the Damköhler number (i.e., the ratio of chemical reaction rate to mass transfer rate) was comparable to what could be expected in a fracture network in the field (on the order of 10−1–10−3).
Shale samples were obtained from Ward's Scientific (Oil Shale #47E7477). CaSiO3 (99%) and CaCO3 (99.95–100.05% dry) were obtained from Sigma-Aldrich. Food-grade liquid CO2 (99.9%) was supplied by Robert's Oxygen. Shale samples were crushed and sieved before use. All other reagents were used as received. Batch studies were used to characterize the rate and extent of carbonation in the presence of shale over a range of formation T/P conditions, whereas flow-through column studies were conducted to quantify porosity and infer permeability for a subset of T/P conditions.
For the batch reaction, solid shale samples were ground using miller jars and sieved to obtain particles with diameters in the range of 37–177 μm. Reactants were packed in a stainless steel reactor (MS-13, HIP, 10 mL) with a mixture of 0.8 g ground shale sample (37–177 μm) and 0.8 g CaSiO3 followed by the injection of 8 g H2O. One end of the reactor was sealed and the other end was connected to the high pressure CO2 supply system controlled by a syringe pump (500HP, Teledyne, Inc.). The reactor was fixed horizontally on a Model 75 Waist Action@ shaker from Burrell, Inc. for continuous shaking. The reactor was placed in an LBB1-43A-1 oven from Despatch, Inc. for constant temperature. The accuracy of temperature is ±0.1°C. Experiments were carried out at two T/P conditions: 55°C/12.4 MPa and 75°C/15.2 MPa. The lower T/P condition was selected to serve as a reference condition for shallower subsurface depths, where hydraulic fracturing is less likely to be deployed (∼1,200 m). The higher T/P correspond approximately with the average depth of the Marcellus shale (∼2,000 m). The morphological and elemental composition changes of mineral samples were characterized before and after reaction using a Quanta 650 scanning electron microscope (SEM) coupled with energy dispersive X-ray (EDS) spectroscopy. Au/Pd was used to coat the sample before SEM/EDS analysis. The sample was fixed on PELCO Tabs™ carbon conductive tabs. EDS measurements were carried out at 15–20 kV.
Changes in the composition of the samples were quantified with X-ray diffraction (XRD) using a PANalytical X'Pert Pro Multipurpose Diffractor unit with Cu-Kα radiation. TiO2 was chosen as the internal reference for its distinguishable Bragg peaks relative to shale and CaCO3. TiO2 was added as a reference to the batch and column experiments as chemically inert means to evaluate the extent of carbonation. Mineral analysis of shale matrix suggests that there is no detectable naturally occurring TiO2 in the shale either before or after it is reacted with high pressure CO2. The diffraction patterns for shale, CaCO3 (calcite), CaSiO3 (wollastonite), TiO2, and the mixture of shale+CaCO3+CaSiO3 are shown in Fig. 2.
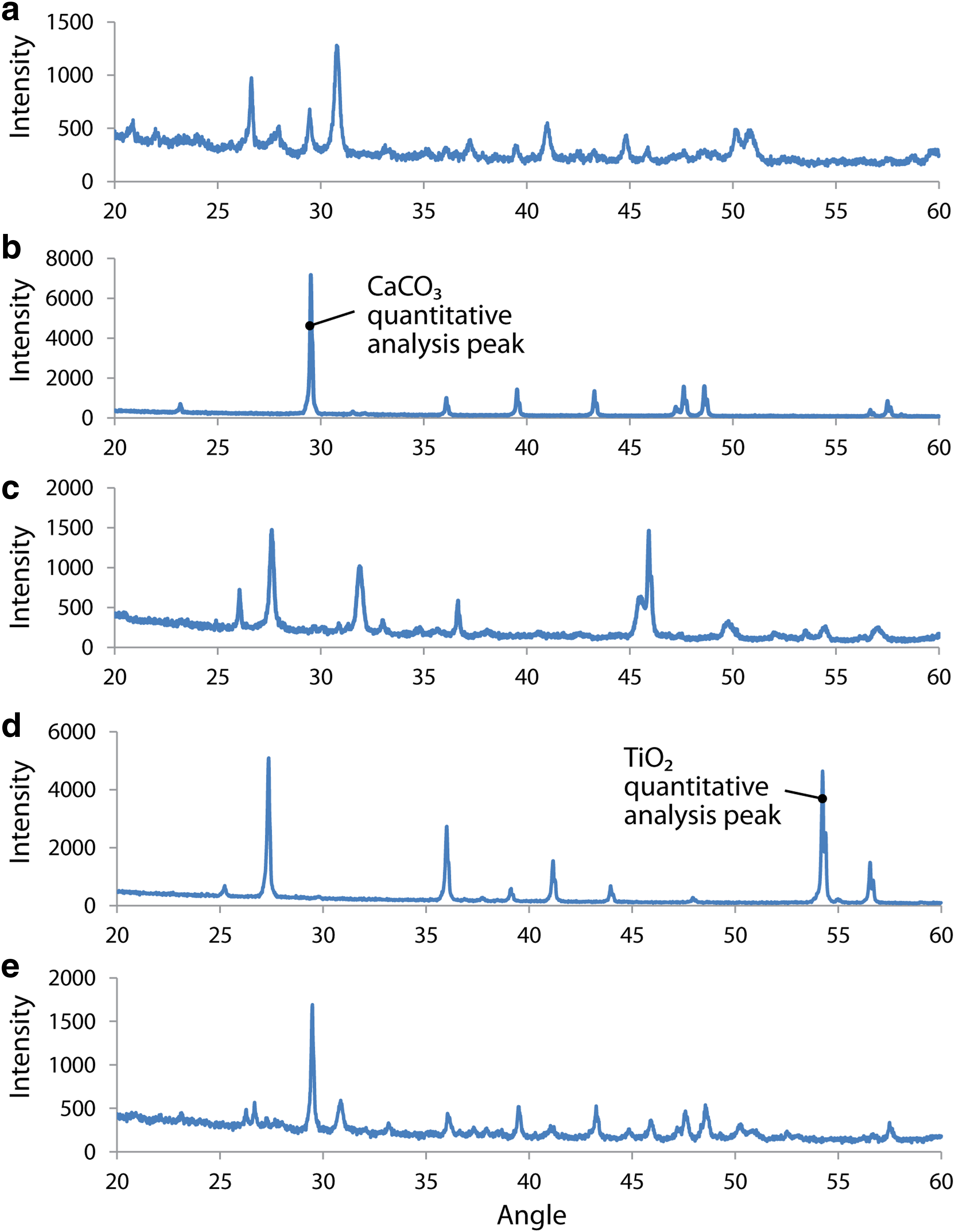
Diffraction patterns for
Ratio between the area of a specific peak from CaCO3 (with arrow in Fig. 2b) and the area of a specific peak from TiO2 (with arrow in Fig. 2d) was used to calculate the amount of CaCO3 produced if the amount of TiO2 reference is known based on Equation (1).
where ICaCO3 is the XRD intensity of the selected peak for CaCO3, ITiO2 is the XRD intensity of the selected peak for TiO2, XCaCO3 is the weight fraction of CaCO3, XTiO2 is the weight fraction of TiO2, and k is an experimental constant.
A standard curve was used to derive the produced CaCO3 mass from the XRD peak intensity. The composition for standard samples and the standard curve are shown in Fig. 3.

Standard curve for quantitative XRD analysis. Markers are measured data points and red line represents linear regression used to translate XRD peak intensity to concentration of reaction products. XRD, X-ray diffraction.
The flow-through experimental setup is shown in Fig. 4. Unlike the batch reactions, in which the shale and wollastonite particles were well mixed, in the column experiments the shale and wollastonite powders were packed into two distinct layers in the column (316 stainless steel, 5 cm length with 0.794 cm OD and 0.635 cm ID) and separated with glass wool to prevent advective transport of wollastonite powders into the shale region of the column. CO2-saturated water was then pumped through the column at high T/P. The pressure within the column was maintained through a back-pressure regulator. Stainless steel porous disks with an average pore size of 2 μm were placed at the top and bottom of the column to prevent outflow of shale grains. These frits were also necessary so that the columns could be tested in the mercury intrusion porisometer without being disturbed. The brine–CO2 mixture was pumped through the column at a flow rate of 0.1 mL/min, which would be representative of fluid flow properties in portions of the fracture network after CO2 injection. Each column experiment was run for 16 h, the time enough to observe measureable differences in carbonation extent between conditions.
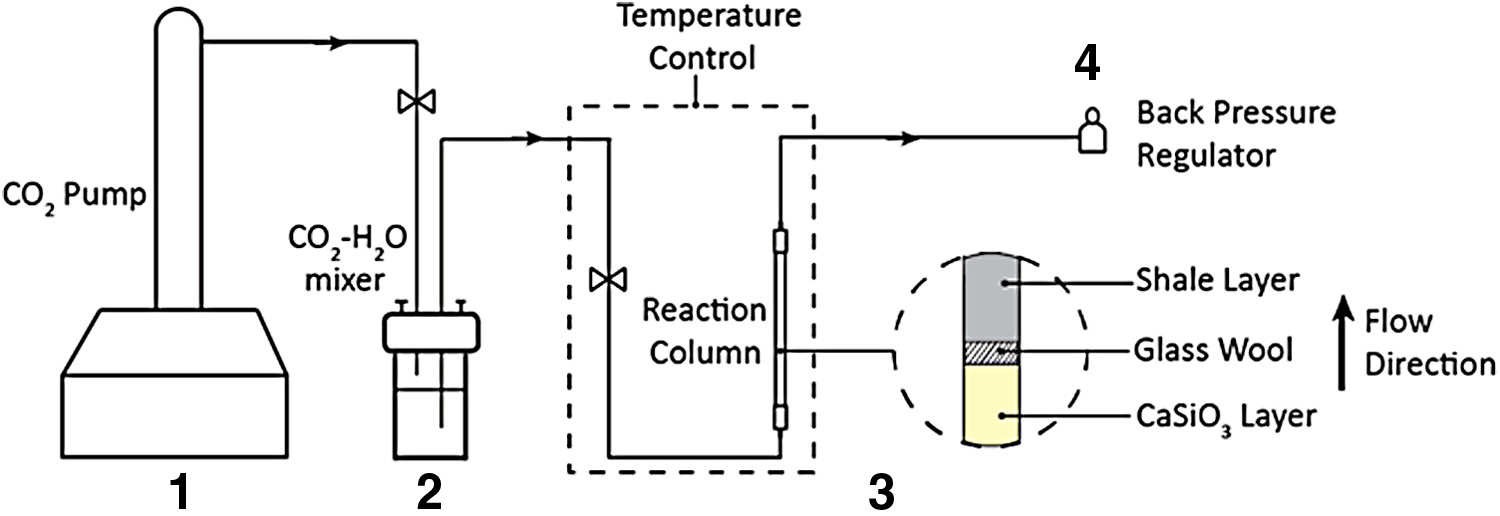
Experiment setup in flow-through configuration. An Isco syringe pump was used to supply food-grade CO2 to a premixer where the brine and CO2 were equilibrated. The reaction column was housed inside an oven to ensure accurate temperature control, and a backpressure regulator was used to maintain a constant pressure within the column.
Mercury intrusion porosimetry (MIP) was used to analyze the effect of carbonation reactions on pore size distribution and porosity (Micromeritics@ Autopore IV). MIP works by gradually increasing the pressure of liquid mercury in contact with a porous solid and measuring the volume of mercury that is required to achieve the set pressure. As pressure increases, smaller and smaller pores are filled. MIP can be used to characterize pores between 2 nm and 400 μm in diameter. Even though most of the porosity within the bulk shale rock has a diameter toward the lower limit of this scale, our interest was in quantifying the effect to the pores between grains not within them and so most of the pores we observed had a diameter on the scale of 10 μm. To provide an accurate measure of changes in permeability within the shale powders, both the wollastonite and the glass wool layers in the column were removed using a high-speed saw before the column was tested using the permeameter.
Figure 5 presents a pore size distribution for a single test under the condition of 12.4 MPa and 55°C. The inset includes all the pore structures with diameters up to 50 μm, but we primarily focused on the pore size range between 0.1 and 20 μm. For each experimental condition, the pore size distribution of a carbonated sample was compared with that of a control sample, in which the wollastonite layer is replaced by nonreactive glass beads while all other conditions were kept constant. This setup eliminates dissolution of the wollastonite and subsequent precipitation, which would impact the pore structure of the packed column. This maximizes the likelihood that any measured differences between the carbonated and control sample are attributed to the carbonation dissolution reactions. The mercury intrusion volume for the particle filtering stainless steel porous disk in the tubular reactor was treated as background and was subtracted from the measurements. No precipitation occurs in the porous disk since it is located upstream of the wollastonite layer. Experiments were conducted in duplicate for each condition and the pore size percentage under each condition is the average value of the duplicate trails.
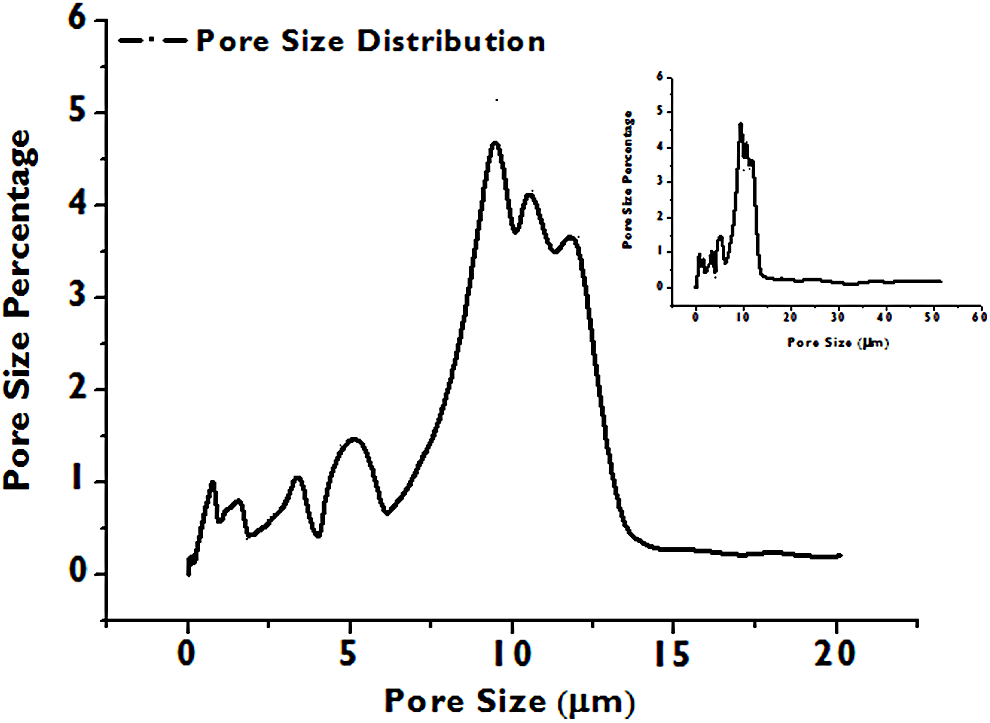
Representative mercury intrusion porosimetry measurement result. Inset includes the pore sizes up to 50 μm indicating that there are few pores with an average size > 20 μm and so our focus was on evaluating changes in pores with average size 0–20 μm.
To translate the porosity data measured experimentally using MIP into permeability estimates, we used two common quantitative relationships. Berg derived a relationship between porosity, particle size, sorting, and permeability: (Berg, 1970)
where k is permeability (mDarcy or 10−15 m2),
Instead of correlating the porosity, particle size, and sorting index with permeability as shown in previous models, Swanson proposed an empirical relationship that used MIP data to derive permeability from more than 300 sandstone and carbonate samples. The permeability is calculated using the following equation:
where k is permeability (Darcy),
Results and Discussion
Batch studies of wollastonite carbonation in presence of shale
Wollastonite was found to react readily with CO2 to form calcite precipitates in the presence of the shale particles at T/P typically encountered in the Marcellus shale formation. XRD patterns for batch experiment reactants and products are shown in Fig. 6. In Fig. 6a, the patterns of the control shale sample before and after reaction are shown to be highly consistent. This suggests that the composition of the shale matrix does not measurably change after reaction with high T/P CO2. The same experimental apparatus is used to carry out reaction with the shale matrix in the presence of CaSiO3, and Fig. 6b shows the XRD patterns are quite different. These scans show a clear increase in the intensity of the peaks that are associated with CaCO3 after the reaction. A correlated decrease in the intensity of the CaSiO3 peak was also observed. The profiles of these scans qualitatively confirm the underlying hypothesis of this work, namely, that mineral carbonation reactions can be carried out in a shale matrix without an antagonistic impact from the shale itself.
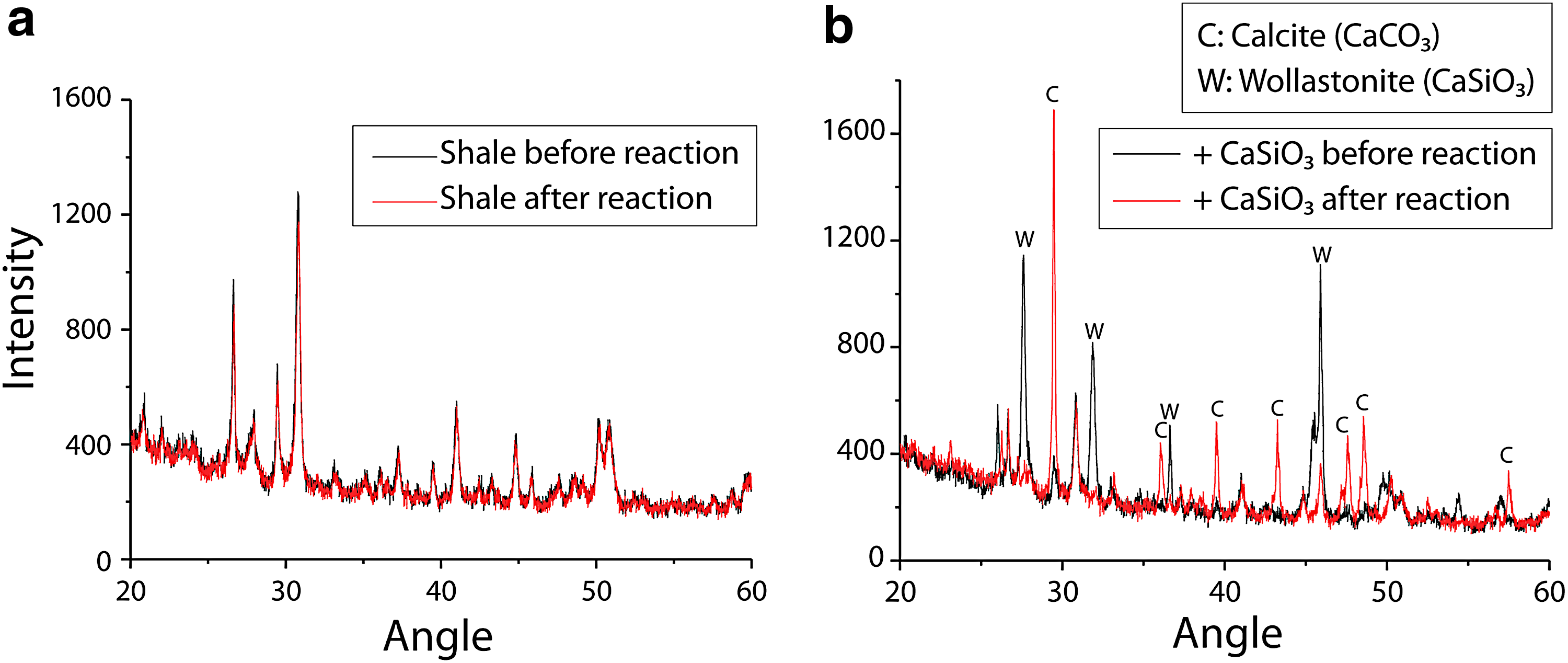
XRD patterns for shale before and after reaction.
Semi-quantitative analyses of the XRD data were carried out to determine the extent of reaction and conversion of wollastonite into calcite under the conditions tested here using an internal TiO2 standard. The results indicate that the reaction achieved greater than 50% conversion (measured in terms of CaCO3 generation) after 24 h. The conversion rate was observed to increase with temperature, which is consistent with published studies of ex situ carbonation (Huijgen et al., 2006).
Spatial distribution and morphology of carbonation products
The electron micrograph in Fig. 7 shows that the shale particles are coated by large crystalline precipitates only after reaction with CO2 in the presence of wollastonite particles at T/P conditions (75°C/15.2 MPa) equivalent to an approximate depth of 2,100 m (Zoback, 2007). A closer look at these surfaces before and after carbonation reveals that this precipitate exhibits both the elemental composition and morphology of calcite. In contrast, Fig. 8 shows the surface of shale particles in the absence of wollastonite before (Fig. 8a) and after (Fig. 8b) reaction. EDS elemental maps (below Fig. 8a, b micrographs) show insignificant changes in the elemental composition of the shale surface before and after exposure to high pressure CO2 in the absence of wollastonite. When wollastonite is present, however, the results were quite different (Fig. 8c, d). Before reaction, the shale particle surface looks like the controls with the exception of the wollastonite particles that can be clearly identified in the silicon EDS map under Fig. 8c. After carbonation (under Fig. 8d), silicon signals are attenuated, while carbon signals are more intense, which is consistent with the EDS fingerprint of calcite. Most definitive is that precipitates displaying calcite's characteristic rhombohedral morphology are only observed after carbonation when wollastonite was present in the starting materials (Fig. 8d).

Electron micrograph of ground shale and wollastonite particles

Electron micrographs of shale surface without wollastonite
In addition to the CaCO3 product, an amorphous silica layer is known to form as a reaction byproduct after dissolution of CaSiO3 (King et al., 2010). Although this phase could have an antagonistic effect by coating reactants, we did not observe significant inhibition of the carbonation process. In this application, amorphous silica could instead coat the shale surface and provide additional sealing, inhibiting liquid/gas migration and the mobilization of hazardous elements within the shale. In practice, time may not be limiting given the relatively long time periods over which these sequestration and sealing processes would continue. But the precise time scales at play would depend on the way in which they are deployed (e.g., to seal a formation at some radius around the wellbore for a slow moving fluid, or to close a fracture during a rupture event).
Impacts of carbonation products on flow in porous media
Column experiments show how carbonation of wollastonite particles can reduce porosity and inhibit flow in an idealized porous media of shale particles. The results from the MIP analysis are shown in Fig. 9. From the MIP analysis shown in Fig. 9a, which was carried out at T/P conditions (55°C/12.4 MPa) equivalent to a depth of around 1,200 m, there is little evidence of a change in pore structure after carbonation. The story is quite different at higher T/P conditions (75°C/15.2 MPa), which are representative of a depth of around 2,100 m. Figure 9b shows an appreciable decrease in pore size after carbonation, compared with the control column, which only differs in that wollastonite is replaced with equivalently sized unreactive glass beads, under the condition of 75°C/15.2 MPa. The width of the shaded gray and red areas corresponds to 1 standard deviation from duplicated samples. Before carbonation, the mean pore diameter is located at 13.4 μm. After carbonation, the average pore diameter is 9.5 μm. This shift of the pore size distribution suggests that the carbonation reactions effectively modify the pore structure, with some of the larger pores being narrowed down as carbonates precipitate within the pore network. The porosity of the sample changed from 0.38 ± 0.06 before carbonation to 0.26 ± 0.02 after carbonation.

MIP result for
These MIP results highlight the importance of T/P on wollastonite carbonation. This sensitivity has been noted in the literature but has new implications for deploying these reactions in the subsurface. Published reports of ex situ carbonation suggest that the optimal range for carbonation of wollastonite occurs between temperatures 100 and 175°C and pressures between 15 and 25 MPa (Huijgen et al., 2006; Gerdemann et al., 2007). Most shale formations that are suitable for hydraulic fracturing exist at depths where T/P conditions are somewhat lower than this, as reflected in the T/P conditions selected here. O'Connor et al. (2005) conclude that the carbonation of wollastonite is less sensitive to PCO2 than that of Mg-bearing silicate minerals, such as olivine. In their work, high conversion of calcium silicate to carbonate could be realized at 100°C even at pressures as low as 4 MPa. Huijgen et al. (2006) suggest that the carbonation of silicate minerals would perform differently in two temperature regimes. Below an optimal temperature, the reaction is rate limited by the dissolution of the silicate. Above that temperature, the carbonation is controlled by the precipitation of carbonate. In their work they show that optimal temperature is dependent on pressure, but the lowest temperature is still considerably higher than 100°C. Both O'Connor and Huijgen's work indicate that the carbonation of CaSiO3 is enhanced at higher temperatures and that the reaction is less sensitive to pressure. The critical point of CO2 (7.4 MPa, 31°C) is important in this context because of the nontrivial increase in molecular density that occurs above this T/P. All of our experiments were carried out well above this T/P, so reactivity is not expected to be impacted by the phase behavior of CO2.
Under these high T/P conditions, water and CO2 can be delivered as either hydrated CO2 or carbonated H2O. As expected, the experiments proceeded to completion much more readily in the carbonated H2O. The dissolution/precipitation reactions are water based and so the reaction would be limited under hydrated CO2 conditions, where water content would be <5% of the total mass of fluid in the pore. Even though water films may form on the pore wall under these conditions, mass transfer effects are likely to come into play. Similarly, the method for introducing the wollastonite powders into the column impacted the extent of carbonation. In these experiments, we tried two different techniques for introducing the silicates into the shale region of the column. In the first, a wollastonite layer was placed above the ground shale powders and separated by a layer of glass wool, with the intention of separating where in the column, the dissolution of the wollastonite and the precipitation of the calcite took place. In the second, the wollastonite particles were mixed with the shale powders before reaction. For the latter case, the carbonation within the column appeared far more extensive. It is worth reiterating here that the columns are ∼5 cm long. This suggests that the precipitation and dissolution reactions occur in proximity to one another. Even though the quantitative impacts of fluid phase and wollastonite injection are not included here, these qualitative observations are consistent with our understanding of the underlying physicochemical processes driving this process.
The porosity measurements were used to estimate the change in permeability in our samples using two empirical relationships. The Berg equation was used to predict the permeability as a function of grain size and porosity. Whereas the Swanson model was used to calculate permeability based on the MIP results directly. The comparison of these methods is shown in Table 1. The results suggest that the two models are relatively consistent and predict an appreciable permeability decrease under the higher temperature conditions tested here. The low P/T sample shows a statistically insignificant reduction in permeability. It is important to note here that Berg's model is particularly sensitive to porosity since porosity is raised to the power of 5. Some of the differences between the estimates, particularly at high P/T conditions, can be attributed to this effect. Compared with the low P/T samples, the permeability decrease of 50% after the carbonation under high P/T suggests that this approach could be deployed in deeper formations where the reaction kinetics would be more favorable.
To calculate the permeability according to Berg's equation, we use the same particle size before and after reaction.
Permeability control in subsurface
To help understand where in the subsurface this approach could be deployed, the chemical kinetics of the underlying precipitation/dissolution reactions was evaluated using first-principal relationships and published data. A heat map describing the results of this analysis across a range of representative geothermal gradients and hydrostatic pressures is presented in Fig. 10. The average properties of several shale formations in the United States are presented as a benchmark. These formations occur generally deeper than 1 km and can occur at different temperatures given the range of lithostatic gradients found in the subsurface. To derive the heat map, the activation energy of the carbonation reaction was calculated at a reference state (25°C, 1 atm) and found to be 36.4 kJ/mol (Daval et al., 2009). We assumed the activation energy was constant with temperature. The reaction rate constant was derived using the Arrhenius equation (Laidler, 1984), as shown in Equation (4):
where kT indicates the reaction rate constant at temperature T. A is the prefactor, Ea is the activation energy, R is the ideal gas constant (8.314 J/[K·mol]), and T is temperature in Kelvin.
Equation (4) can be modified to calculate the change in the reaction rate constant from temperature T0 to T through Equation (5):
As discussed earlier, the rates of the carbonation of wollastonite are less dependent on pressure than temperature. Even though CO2 concentrations would increase significantly above the critical pressure, the pressure conditions reported here are far from the critical point. Huijgen et al. (2006) reports a modest increase of carbonation reaction rate from 2 to 4 MPa at 100°C. Gerdemann et al. (2007) indicates that the reaction rate of carbonation reaction for different minerals is weakly related to pressure change (compared with temperature) and it is projected that the increase of reaction rate from the critical pressure of CO2 (7.4 MPa) to the highest pressure in our range (40 MPa) is on the order of one to three times. To capture the effect of pressure, we assume that the reaction rate constant would double over the pressure range evaluated here at a constant temperature.
Heat map results presented in Fig. 10 are expressed in terms of factor increase in reaction rate constant relative to the reference state at the critical point. The reaction rate constant increases with T/P as indicated by the transition from blue to yellow to red. The color bar at the right side indicates how many times the rate constant changes compared with the bechmark value of k at the CO2 critical point (illustrated here in the upper left hand corner of the figure). The maximum point occurs at the bottom right hand corner of the figure. It is worth noting that the carbonation of Mg-bearing silicates (analysis not included here) would generally require higher T/P (O'Connor et al., 2005) than the Ca-bearing silicates modeled here. So although calcium silicates like wollastonite would be suitable for shallower formations, magnesium silicates would be more suitable for deeper formations.

Reaction rate of carbonation of calcium silicate in subsurface as a function of depth, temperature, and pressure. Phase behavior of CO2 and the approximate conditions in three of the major shale plays in the United States (the Marcellus, Barnett, and Haynesville) are also presented. The two lines represent a characteristic lower and upper bound on the thermal gradient in the subsurface. Colors represent the increase in reaction rate relative to a reference state (at the critical point).
Environmental Implications
This work suggests that mineral carbonation of wollastonite can be carried out effectively in the presence of shale under the T/P conditions representative of the Marcellus formation. The results are expected to apply generally to many other shale plays at similar or greater depths, where conditions will promote in situ mineral carbonation. Shale-fracturing activities could lead to a variety of environmental risks and these mineral carbonation reactions could have important implications for developing strategies for reducing the hydraulic connectivity of disturbed shale formations during the postclosure phase of a well life cycle. A further understanding of how these in situ carbonation strategies impact flow will require bench-scale studies on fractured shale cores and field trials, but the morphology of the precipitated CaCO3 and its association with the shale matrix surface suggest that this process would reduce mesoscale and macroscale fluid flow within the shale matrix and fracture networks alike. The proposed process could also play an important role in efforts to re-engineer depleted shale plays as secure CO2 storage reservoirs and/or impervious caprock seals for deeper saline aquifers.
Even though the experiments reported here are for one representative mineral carbonation reaction, it is likely that similar reactions, such as those containing Mg-bearing species, would behave in much the same way. It is worth noting that the presence of CH4 and other hydrocarbons is not expected to trigger the carbonation of silicate minerals, although there are ways in which the presence of hydrocarbons could impact the deployment of this technology. For example, if the silicate minerals are covered by a layer of hydrocarbons, this could suppress the dissolution by CO2 and impact overall reaction kinetics.
In addition to shale fracturing, there are other emerging energy technologies that are being deployed in the deep subsurface where selective permeability control would be desirable. Carbon capture and sequestration activities in saline aquifers are being studied as a means of permanent storage of CO2, and techniques for sealing leaks or maintaining wellbore integrity are of great interest. In another example, fluid injection associated with produced water disposal is contributing to seismicity that is of great concern to the oil and gas industry. Some of these risks could be potentially mitigated if target formations were cemented during or after fluid injection. Finally, fluid or gas storage in the deep subsurface would be an effective means of energy storage as long as risks from leakage could be prevented.
Recent work by Kang et al. (2014) and others suggests that the legacy risks from gas wells account for a significant amount of greenhouse gas emissions to the atmosphere. These emissions are generally not accounted for in many climate models and could contribute to the underestimation in carbon budgets globally. Most work on abandoned wells suggests that leaks around wellbores are due to faulty construction and/or wellbore integrity failures over decades and centuries after closure of the well. A method to stabilize the entire formation, or at a minimum the region of the formation closest to the wellbore, could provide a much more robust strategy for stabilizing geologic formations that have been disturbed during hydrocarbon extraction. In an increasingly carbon-constrained economy, such methods for mitigating leakage and seepage of gases into the atmosphere will be critical.
Conceptually, these results suggest that this method could be used to manage many of the legacy risks associated with hydraulic fracturing. In practice, methods will be needed to deploy this technique in the field. One approach to deploying this technology would be to substitute CaSiO3 proppants for sand (as described in Fig. 1), during the initial fracturing of the gas-bearing zone and then once gas production is complete, CO2 injection would initiate carbonation reactions to seal the formation. Such an approach would depend on the gas and water composition to prevent unwanted decrease in permeability while it is still in the production phase. In other applications, such as geologic carbon sequestration, the calcium or magnesium silicates might be injected with coatings to limit their reactivity to only regions where flow is undesirable, such fractures where leaks from a target repository may be occurring. This article suggests that the technique could form an enabling method for controlling the properties of porous media in a range of applications in the deep subsurface.
The use of mineral carbonation reactions could have other applications for wells and shale formations in the long term that could be a challenge to foresee today. For example, this technique could make it easier to drill wells closer to each other because risks of connectivity between fracture networks would be reduced. Conversely, if new technology is developed decades from now that would enable well operators to reopen old wells and extract natural gas that is currently inaccessible, this technique might confound such efforts. Given that this technology is in the proof-of-concept stage, efforts to understand these long-term implications are highly uncertain but should be considered as the technology is transferred to the field.
Conclusions
This article presents proof-of-concept results that suggest that carbonation of introduced mineral silicates could be used as a strategy to decrease fluid connectivity and cement porous formations in the deep subsurface. In circumstances where decreasing porosity and permeability is desirable, this approach could help mitigate seepage and other related environmental risks. The experiments reported here support a number of findings:
• Mineral silicates mixed with dense phase CO2 and water will produce solid carbonate precipitates that effectively cement and reduce the permeability of unconsolidated shale particles. • Carbonation of mineral silicates proceeds spontaneously under the T/P conditions of many deep subsurface formations. • Shales tested here do not appear to contain minerals or dissolution products that would have antagonistic or synergistic impacts on these reactions. We recognize that the geochemistry of shale formations is very diverse, and for those that contain higher concentrations of carbonates or clays, this class of reactions may be less appropriate because of intrinsic reactivity with CO2. • Reaction kinetics are relatively rapid and occur over the time scale of days. • Reactions proceed much faster in carbonated water than in hydrated CO2. • Dissolution and precipitation reactions occur in spatial proximity, so the injection of concentrated cation solutions with CO2 would decrease permeability near the injection site. Injection of cation donor-bearing mineral silicates into the formation followed by subsequent injection of a CO2-rich aqueous phase will result in better spatial distribution of carbonation precipitates throughout the target formation. • An obvious amorphous silica layer also precipitates and this could impact fluid flow. • Carbonation reactions would decrease porosity and permeability of a fractured media.
Footnotes
Acknowledgments
This work is supported by the U.S. National Science Foundation Awards # CBET-1134397 and CBET-1254839 and U.S. Department of Energy Award # DE-FE0026582.
Author Disclosure Statement
No competing financial interests exist.