
Abstract
Abstract
Electrochemical degradation of 2,4-dichlorophenol (2,4-DCP) in aqueous solution has been studied by a dual-cathode oxidation system (DOS), in which a gas-diffusion electrode was used to produce H2O2 in situ by oxygen reduction, a graphite cathode was employed to accelerate the reduction of Fe3+ and a Ti/SnO2-Sb2O5-IrO2 anode was used for the anodic oxidation (AO) of organics. It is shown that both electro-Fenton oxidation and AO account for the degradation of 2,4-DCP in this process. Comparative degradation showed that DOS could present a more rapid removal of 2,4-DCP compared with the single-cathode oxidation system. When −0.70 and −0.10 V were applied to the gas-diffusion cathode and the graphite cathode, respectively, the degradation of 2,4-DCP solution (100 mg/L) with 0.15 mM Fe2+ at pH 3.0 for 240 min yielded 80% total organic carbon removal and a mineralization current efficiency of 20%. The effect of DOS treatment on the toxicity reduction was evaluated by the dehydrogenase activity (DHA) of activated sludge. It was found that DHA value became greater via the treatment in contrast with the DHA value of untreated solution. This work suggests that DOS might present an alternative for the treatment or the pretreatment of effluents containing chlorophenols.
Introduction
C
In recent years, electrochemical oxidation has been considered as one of the most promising technologies that is environmental friendly and can oxidize toxic and bio-refractory organics effectively (Martinez-Huitle and Ferro, 2006; Brillas et al., 2009; Rodrigues de Oliveira et al., 2011). Electrochemical oxidation processes mainly include (i) anodic oxidation (AO) characterized by the generation of ·OH via water oxidation at the surface of a high O2-overvoltage anode from reaction (1) and (ii) indirect electro-oxidation method with H2O2 electro-generation via O2 reduction from reaction (2), so-called electro-Fenton oxidation (EFO) using Fe2+ ions as catalyst generating ·OH by reaction (3) (Comninellis, 1994; Brillas et al., 2009; Babuponnusami and Muthukumar, 2012):
where M (·OH) denotes the hydroxyl radical adsorbed on M (anode) or remained near its surface. In the past two decades, many studies have reported the application of AO for organic wastewater remediation by the various anodes such as Pt, IrO2, RuO2, SnO2, PbO2, and boron-doped diamond (BDD) (Martinez-Huitle and Ferro, 2006; Panizza and Cerisola, 2009). However, industrial applications of AO are still limited due to the relatively high energy consumption or the high electrode price.
As a popular indirect electro-oxidation method, EFO has been applied to treat many industrial wastewaters containing phenol (Pimentel et al., 2008), aniline (Brillas and Casado, 2002), herbicides (Badellino et al., 2007), dyes (Ruiz et al., 2011), and so on. In electro-Fenton process, reaction (3) can be propagated from Fe2+ regeneration by the reduction of Fe3+ at the cathode and/or the reduction function of several species such as H2O2, HO2
Gas-diffusion electrode (GDE) is the most popular cathode used in EFO characterized by its superiority in H2O2 generation. At this cathode, the reaction (2) is predominant giving rise to a high H2O2 production, while reaction (4) is very slow and most iron species are in the form of Fe3+. However, based on many studies, some small molecule organic acids are always presented during organics degradation by Fenton or electro-Fenton process (Dirany et al., 2010), and these carboxylic acids can react with Fe3+ generating complexes. Because it is very difficult to destroy these Fe(III) complexes by ·OH generated in electro-Fenton process (Guinea et al., 2008; Skoumal et al., 2008), the significant decrease of degradation efficiency was obtained (Anotai et al., 2006; Trovó et al., 2011). Moreover, the drawback of conventional electro-Fenton process is that it easily produces iron sludge, and it is difficult to deal with this iron sludge after wastewater treatment. It is evident that the low dosage of Fe2+ will be beneficial to reduce the amount of iron sludge. Therefore, some effort to reduce the generation of Fe(III) complexes and the amount of iron sludge by reducing the initial Fe2+ concentration should be concerned.
In electro-Fenton process, if H2O2 is electrogenerated on cathode surface under the optimum conditions, simultaneously, the AO of organic contaminants is also used by the greatest extent, the purposes of enhancing current efficiency and reducing energy consumption will be realized. However, from an economical point of view, since electrochemical oxidation process is fairly expensive in comparison with biological treatment, the former may be used as an auxiliary unit to enhance the biodegradability of effluents followed by biological treatment. It has been reported that the biodegradability of textile dye wastewater could be improved evidently after treatment by the AO of Ti/Pt (Vlyssides et al., 1999). Nevertheless, until now little work was conducted to enhance the biodegradability of chlorophenols solution by reducing its toxicity via EFO coupled with AO process.
This article reported a novel dual-cathode oxidation system (DOS) using Ti/SnO2-Sb2O5-IrO2 as anode, gas diffusion electrode as the first cathode for H2O2 generation, and graphite electrode as the second cathode for Fe2+ regeneration. The aims of this work were to investigate the effectiveness and the characteristics of 2,4-dichlorophenol (2,4-DCP) degradation by DOS under the low dosage of Fe2+, and learn about whether DOS can reduce the toxicity of 2,4-DCP solution.
Materials and Methods
Chemicals and electrodes
All chemicals were analytical grade reagents and used as received. All solutions were prepared with deionized water (conductivity <1 μS/cm). The supporting electrolyte consisted of a 0.10 mol/L Na2SO4 solution and 2,4-DCP was added to this solution used as the simulated pollutant. The pH value of all electrolytes was adjusted to 3.0 using 10% H2SO4 solution.
Ti/SnO2-Sb2O5-IrO2 anode was prepared by the thermal decomposition method (Chu et al., 2010). The graphite electrode with a carbon content of 99.5% was used as the second cathode. GDE was prepared by the following steps.
Graphite powder (99.9% purity) and polytetrafluoroethylene (PTFE) emulsion were mixed with a mass ratio of 3:1 (Wcarbon:WPTFE = 30:10 g), followed by the addition of 10 mL NH4HCO3 (200 g/L) and 15 mL ethanol. After the above mixture was well mixed by ultrasonic equipment, it was shaped into the cylinder with a diameter of 24 mm using an extruder, and then was dried at 330°C for 60 min in a muffle furnace. After that, the prepared material was dipped into acetone for 24 h. Finally, it was dried at 100°C for 60 min.
Electrolytic system
The electrolytic experiments were performed in a 250 mL beaker containing 200 mL solution with a magnetic bar. The potential values of GDE and graphite cathode were constantly controlled by a bi-potentiostat (CHI760D Electrochemical Workstation purchased from Shanghai, China). The GDE was fed with an air flow rate of 25 mL/s to continuously electrogenerate H2O2 from O2 reduction. The graphite cathode was used for the reduction of Fe3+ in DOS, but disabled in single-cathode oxidation system (SOS). The geometric area in contact with the electrolyte was 38 cm2 for the anode, 30 cm2 for GDE, and 38 cm2 (or 19 cm2) for the graphite cathode. The distance between anode and cathode was 15 mm. In all electrolytic experiments, a saturated calomel electrode (SCE) was used as a reference electrode.
Electrochemical experiments
Degradation of 2,4-DCP was carried out in the electrolytic system described in “Electrolytic System” section. For each run, 200 mL of the solution containing 2,4-DCP and 0.10 mol/L Na2SO4 (pH 3.0) was poured into the reactor. For the different experimental purposes, FeSO4·7H2O with different dosage was added into the solution. The potential of GDE was controlled at the desired value and the potential of graphite cathode was controlled at −0.10 V/SCE in DOS. At appropriate time intervals, samples were taken for chemical analysis. Before each experiment, GDE was subjected to a hot treatment at 330°C for 60 min, and then washed by deionized water to obtain a stable H2O2 production. All experiments were performed at least twice at 22°C ± 2°C.
Dehydrogenase activity investigation of activated sludge
Activated sludge presented by a municipal sewage treatment plant was aerated with air for 360 min, and then it was condensed to a solid concentration of 9,000 mg/L via gravity settlement. After the electrochemical treatment, the solution was adjusted to 7.0 of pH value followed by the filtration with filter paper. A small amount of Na2SO3 (30 mg/L) was added into the above solution. This solution of 300 mL was poured into a 500 mL beaker, followed by the addition of 100 mL activated sludge, and then aeration was presented to supply O2. The mixed liquor of 20 mL was withdrawn for dehydrogenase activity (DHA) determination at 0, 60, 120, 240, 360, 480, and 600 min, respectively.
Analytical methods
Before the quantification of 2,4-DCP, samples were subjected to distillation pretreatment, and then the concentration was achieved by 4-amino antipyrine spectrophotometric method at 510 nm. Total organic carbon (TOC) was measured using TOC-VCPH Analyzer (Shimadzu). The quantification of H2O2 and Fe2+ was performed immediately after samples were withdrawn from treated solutions. H2O2 concentration was determined by the light absorption of the titanium-hydrogen peroxide colored complex at λ = 410 nm (Chu et al., 2013). Fe2+ was determined by 1,10-phenanthroline spectrophotometric method (Chu et al., 2013). Cl− was determined by a Dionex ICS-3000 ion chromatograph system. Oxalic acid was measured by ion exclusion chromatography with a Supelcogel H column at 40°C. The detection was conducted at 210 nm with a mobile phase of 4 mM H2SO4 at a flow rate of 0.25 mL/min. Additionally, ultraviolet (UV)-vis adsorption spectra were recorded using a TU-1901 spctrophotometer (Beijing Puxi Co. Ltd.). DHA was determined by the colorimetric method based on the reduction of 2,3,5-triphenyl tetrazolium chloride (TTC) generating triphenyl formazan (TF) (Chu et al., 2015).
Results and Discussion
Degradation of 2,4-DCP in absence of Fe2+
Electrochemical degradation of 2,4-DCP in the absence of Fe2+ was conducted by SOS to explore the AO at Ti/SnO2-Sb2O5-IrO2 and the reasonable cathodic potential for H2O2 electrogeneration at GDE. Fig. 1a shows the decay of 2,4-DCP concentration under various cathodic potential values. The more negative cathodic potential accompanied with a more power AO because of the higher anodic potential and current, led to the relatively rapid concentration decay. When without potential externally imposed on electrodes, the concentration decay is due to the volatilization of 2,4-DCP. It is obvious that the volatilization can enhance the elimination of 2,4-DCP from the solution during electrochemical degradation. But it should be noted that the degradation of 2,4-DCP can weaken the volatilization for the dynamics reason.

Decay of 2,4-DCP concentration
As clearly seen from Fig. 1b, the GDE at −0.70 V exhibited a higher catalytic activity toward O2 reduction generating H2O2. In this case, H2O2 accumulation of 75 mg/L was achieved by the electrolysis of 90 min, much greater than those obtained at −0.50 and −0.90 V. Additionally, it should be pointed that the real H2O2 production was more than the measured production because of the H2O2 decomposition caused by several processes (Drogui et al., 2001; Agladze et al., 2007). As the cathodic potential is −0.90 V, the current increases obviously because of H2 evolution by H+ reduction, leading to the decrease in H2O2 accumulation. Therefore, in the following experiments, the potential of GDE was constantly controlled at −0.70 V.
Effect of Fe2+ on 2,4-DCP degradation by DOS
A series of experiments were performed to examine the effect of Fe2+ on 2,4-DCP degradation by DOS using −0.70 V (average current of 75 mA) for GDE and −0.10 V (average current of 7 mA) for graphite cathode. The concentration decay with various Fe2+ concentrations is shown in Fig. 2a. It is apparent that Fe2+ plays a key role in the process. Since the single oxidation of H2O2 is unable to destroy 2,4-DCP, the degradation is mainly ascribed to the combination of EFO and AO in the presence of Fe2+. A significant decay was also attained in the absence of Fe2+ (0 mM) owing to the AO at Ti/SnO2-Sb2O5-IrO2. The degradation mechanism of organics by this kind of anode has been proposed by several researchers (Comninellis, 1994; Martinez-Huitle and Ferro, 2006). It can be observed that the increase of initial Fe2+ concentration from 0 ∼ 0.15 mM greatly enhances the decay of 2,4-DCP. Compared with the decay at 0.15 mM, an approximate or a slightly slower decay was obtained with 0.20 mM. Therefore, excess Fe2+ does not result in the faster degradation of 2,4-DCP, which may be explained by the quenching effect of ·OH caused by Fe2+ from the reaction shown in Equation (5).
Considering the removal of 2,4-DCP and the minimization of Fe2+ dosage, the optimum concentration for an effective degradation is 0.15 mM. This result can be confirmed by the fact that the maximum TOC removal is favored by 0.15 mM Fe2+ (Fig. 2b). Additionally, although the complete removal of 2,4-DCP was achieved by the electrolysis of 90 min with 0.15 mM Fe2+, only 40% TOC could be removed from the solution indicating that part of 2,4-DCP was degraded to soluble intermediates rather than completely oxidized to CO2 and H2O.

Effect of initial Fe2+ concentration on 2,4-DCP decay
Comparative degradation by SOS and DOS
A comparative study on the degradation of 2,4-DCP solution was conducted by SOS and DOS. For the purpose of comparison, the potential of GDE was controlled at −0.70 V in the two processes to obtain an approximate H2O2 production. The concentration decay curves obtained with 0.10 and 0.15 mM Fe2+ are shown in Fig. 3a and b, respectively. Clearly, DOS can present a more rapid degradation than SOS and a relatively longer treatment time is required for the latter to destruct all 2,4-DCP molecules. The mineralization of 2,4-DCP is expected to release its Cl atoms in the form of Cl−. Figure 3c shows the variation of Cl− content during the degradation by the two processes with 0.15 mM Fe2+. The faster dechlorination rate from DOS further confirms that it can give rise to the faster degradation of 2,4-DCP than SOS.

Comparative degradation of 2,4-DCP by DOS and SOS using 0.10 mM Fe2+
In above experiments, the average anodic current values were 83 mA for DOS and 81 mA for SOS and this slight difference in anodic current demonstrated that the noticeable difference of degradation rate was not caused by the different AO power. Therefore, the relatively faster degradation by DOS might be due to the presence of graphite cathode accelerating the regeneration of Fe2+, generating more hydroxyl radicals for organics degradation. During Fenton reaction, Fe2+ is consumed more rapidly than it is regenerated (Duesterberg et al., 2008; Masomboon et al., 2009) and its regeneration plays a key role in the lasting of Fenton reaction. Since the main function of GDE is the electrogeneration of H2O2, the regeneration of Fe2+ by the cathodic reduction in SOS is very weak, whereas the regeneration of Fe2+ can be enhanced by the reduction at graphite cathode in DOS. Compared with SOS, DOS can lead to the rapid change between the Fe(III)/Fe(II) couple resulting in the well degradation even with a lower initial Fe2+ concentration.
The typical evolution of Fe2+ concentration during the degradation by DOS and SOS is shown in Fig. 3d. Note that similar trend can be found operating with the initial Fe2+ concentration of 0.10 or 0.15 mM. It is clear that the concentrations obtained from DOS are always greater than those from SOS, which further evidences the function of Fe3+ reduction at the graphite cathode.
Degradation performance and toxicity reduction by DOS
The experiments were carried out at −0.70 V for GDE and −0.10 V for graphite cathode using 0.15 mM Fe2+ with a longer electrolysis time to learn more about the degradation by DOS. TOC removal exhibited a rapid increase as electrolysis time was increased from 0 to 30 min, followed by an approximately linear increase, and 80% TOC removal was achieved by the electrolysis of 240 min indicating a well mineralization degree (Fig. 4a). Additionally, as the treatment proceeded, the solution changed from colorless to orange-yellow and then gradually disappeared with prolonged reaction time, which demonstrated visually that some intermediates were formed and destroyed during the degradation.
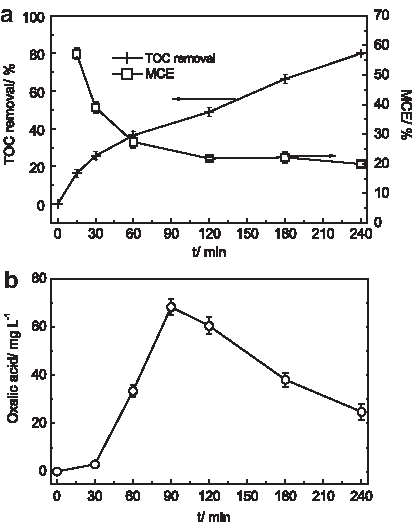
TOC removal and MCE as a function of reaction time
Mineralization current efficiency (MCE) is an important parameter to evaluate the effectiveness of electrochemical processes. MCE at a given time t (h) was calculated using Equation (6) (Skoumal et al., 2009). Where F is the Faraday constant (96,487 C/mol), V is the solution volume (L), Δ (TOC)
t
is the TOC decay (mg/L), 4.32 × 107 is the conversion factor for units homogenization (3,600 s/h × 12,000 mg/mol), m is the number of carbon atoms of 2,4-DCP (6 C atoms), and I is the applied total current (A). The number n of electrons consumed per 2,4-DCP molecule during mineralization was taken as 24 considering that it is converted into CO2 and Cl− ions according to the following reaction [Equation (7)].
A MCE value of 55% was obtained at 30 min, which decreased to 20% at the end of the electrolysis (Fig. 4a). The higher MCE values were observed at the early stage of the electrolysis indicating the fast conversion of organics into CO2, followed by a deceleration at long electrolysis times as a consequence of the loss of organic matter and the formation of more difficultly oxidizable organic intermediates.
During the degradation of 2,4-DCP by DOS, oxalic acid concentration increases rapidly from 0 to 90 min, followed by a noticeable decay due to its further degradation (Fig. 4b). Although oxalic acid was not the only intermediate, it was the major intermediate generated during the degradation, which could be inferred by comparing the concentration values of oxalic acid and TOC. As we all know, oxalic acid has a high biodegradability compared with 2,4-DCP, hence it may be feasible that using DOS as a pretreatment to reduce the toxicity enhancing the biodegradability of the solution.
Figure 5 shows the content of 2,4-DCP and UV-vis spectra changes of the solutions treated by DOS with various reaction times. A main absorption band for 2,4-DCP was at 260 nm and the absorption intensity reflected its concentration in the solution. The peak intensity decreased with the reducing of 2,4-DCP confirming the rapid degradation in this system. The absorption peak disappeared after 180 min indicating a complete destruction of the molecular structure of 2,4-DCP.
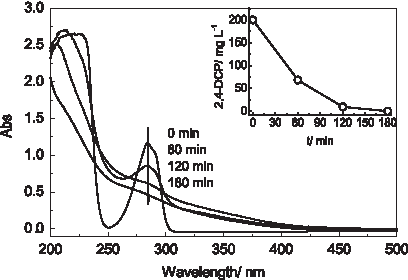
Change of UV-vis absorption spectra (samples were diluted two times) and 2,4-DCP decay with reaction time obtained from the degradation of 200 mg/L 2,4-DCP solution. The potential values of GDE and graphite cathode were controlled at −0.70 and −0.10 V, respectively. The initial concentration of Fe2+ was 0.15 mM. UV, ultraviolet.
Results of MCE indicate that the application of this process in treating the waste waters containing 2,4-DCP at large scale may be limited by the high power consumption. However, it may be an alternative to couple the versatility of DOS with the cheapness of biological methods by the following process: DOS can be employed to destruct the molecular structure of 2,4-DCP reducing the toxicity, followed by biological processes to further reduce organic substances. In this work, the toxicity of 2,4-DCP solution (200 mg/L) before and after the treatment by DOS was evaluated using DHA of activated sludge.
DHA has been widely used to test the toxicity of wastewaters or soils (Diamantino et al., 2001). In this work, the toxicity was evaluated by the DHA of activated sludge. Two samples were prepared by the electrochemical treatment of 200 mg/L solution at −1.0 V with 0.15 mM Fe2+ for 60 and 120 min, respectively. Additionally, one untreated sample was provided for DHA analysis as a control group.
Figure 6 shows that the contact time of activated sludge in the solution has a tremendous effect on DHA values. For the untreated solution, DHA rapidly decreases to 20.2 from 325.0 mg TF (L· h)−1 in 120 min, and finally decreases to 2.9 mg TF (L· h)−1 at 600 min. This result shows that the raw sample of 200 mg/L 2,4-DCP solution has a higher toxicity and can pose a strong inhibition on microbial activity. In contrast, DHA decreases to 106.8 and 211.5 TF (L·h) −1 in 120 min obtained from the samples treated by DOS for 60 and 120 min, respectively. More importantly, DHA does not decrease continuously, but increases after 360 min, which exhibits that the microorganism contained in activated sludge gradually adapt to the solution environment. From above results, it can be concluded that DOS process has reduced the toxicity of 2,4-DCP solution evidently. The reason of reducing toxicity may involve the destruction of benzene ring and de-chlorination generating some biodegradable intermediates. According to this result, it may be feasible to use DOS coupled with biological processes for the treatment of those waste waters containing the high content of chlorophenols.
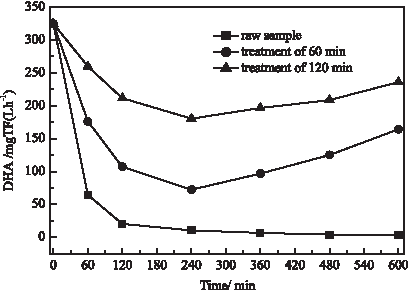
Effect of DOS treatment on DHA of activated sludge. Potential values of GDE and graphite cathode were controlled at −0.70 and −0.10 V, respectively. The initial concentration of Fe2+ was 0.15 mM. DHA, dehydrogenase activity.
Conclusions
In this study, the effectiveness of 2,4-DCP degradation by a DOS involving AO and EFO was found. This process presented a more rapid degradation than the SOS, due to the presence of graphite cathode accelerating the reduction of Fe3+ with a low initial concentration of Fe2+. Under the optimum potential conditions: −0.70 V for GDE and −0.10 V for graphite cathode, the degradation of 100 mg/L 2,4-DCP solution with 0.15 mM Fe2+ at pH 3.0 for 240 min yielded 80% TOC removal accompanied by a MCE of 20%. According to these results, it is feasible to use DOS for the treatment of wastewaters with low content. The variation of activated sludge DHA demonstrated that DOS treatment was able to remarkably reduce the toxicity of 2,4-DCP solution. Therefore, the combination of DOS and biological processes might present an alternative to deal with the wastewaters containing high content 2,4-DCP.
Footnotes
Acknowledgment
This research was funded by the National Natural Science Foundation of Shandong Province under Grant No. ZR2014EEM032.
Author Disclosure Statement
No competing financial interests exist.