
Abstract
Abstract
This article reports on the application of an advanced reduction process (ARP) that combines ultraviolet (UV) irradiation and dithionite to remove selenium from solution. Batch kinetic removal of selenite (Se(IV)) and selenate (Se(VI)) with the dithionite/UV ARP at different pH were evaluated under anaerobic conditions and a removal mechanism was proposed. Resolubilization of selenium was observed for all pH (7, 8, and 9) after 15 to 20 min, which was the time when dithionite was completely consumed. A hypothesis was proposed that selenium first was reduced to elemental Se by radicals formed from photolysis of dithionite. Then, it was resolubilized by reaction with oxidizing radicals formed by photolysis of dithionite degradation products. A kinetic model for dithionite photolysis and a second-order model for precipitation of selenium were developed to describe selenium removal. Experimental data were used in nonlinear regressions to estimate quantum yields and second-order rate constants. Lowest quantum yields were observed at pH 8 for experiments with both selenite and selenate. The rate of selenite removal from solution was not affected by pH, but the rate of selenate removal was faster at pH 8 than at pH 7 and 9.
Introduction
A
Selenium is a naturally occurring trace element that is found in high-sulfur coals due to its similar chemical behavior as sulfur (Lenz and Lens, 2009). It can be an essential nutrient to humans, but it also can be toxic to humans at higher levels. Dietary deficiency (<40 μg/day) can cause Keshan disease in humans, which is a disease of heart muscle (Lenz and Lens, 2009; Gibson et al., 2012). Excessive selenium (>400 μg/day) increases the risk for dermal and neurological diseases, including loss of hair, weakened nails, skin lesions, and changes in peripheral nerves (Fawell and Nieuwenhuijsen, 2003; Lenz and Lens, 2009; Gibson et al., 2012). Therefore, it is important to regulate human supplementation of selenium. The U.S. Environmental Protection Agency (USEPA) set the maximum contaminant level of selenium at 50 μg/L for drinking water (Environmental Protection Agency (US), 2009). The World Health Organization (WHO) recommended a lower limit of 10 μg/L for drinking water (Bleiman and Mishael, 2010; Gibson et al., 2012; Awual et al., 2015).
A variety of treatment technologies, including ion exchange (Nishimura et al., 2007), adsorption (Su et al., 2008; Wasewar et al., 2009; Bleiman and Mishael, 2010; Han et al., 2013), coagulation-precipitation (Kapoor et al., 1995), membrane processes (Kapoor et al., 1995), and biological treatment (Zhang et al., 2008; Lenz and Lens, 2009) have been applied to remove selenium from water. Most of these techniques for selenium removal depend on the oxidation state of selenium and require low pH conditions (Ling et al., 2015; Jung et al., 2016). Competing anions are the most challenging limitation on removal of selenium with ion exchange resins, adsorption, and enzymatic reduction in biological treatment (Nishimura et al., 2007; Lenz and Lens, 2009). Effectiveness of removal of selenium by ion exchange resins and coagulation-precipitation processes is different for selenite and selenate (Bleiman and Mishael, 2010; Zelmanov and Semiat, 2013). Production of secondary wastes and high operating and maintenance costs inhibit the wide application of precipitation (Lenz and Lens, 2009; Awual et al., 2015). Membrane processes are effective technologies for removing selenium, but their operational costs are high (Kapoor et al., 1995; Bleiman and Mishael, 2010; Zelmanov and Semiat, 2013).
Sodium dithionite is a strong reductant and is known to be able to reduce Se(IV) rapidly by itself (Geoffroy and Demopoulos, 2009; Duan et al., 2017). However, application of UV irradiation increases the removal rates due to more rapid formation of highly reactive reducing radicals through dithionite photolysis (Duan et al., 2017). Photolysis of dithionite is usually described as occurring due to breaking of the weak S–S bond in dithionite and formation of two sulfur dioxide radicals, as shown in Equation (1). The free radicals are able to react with selenium and convert it to solids that are effectively removed by filtration (Duan et al., 2017).
An alternative to this photolysis mechanism is based on reports of a dithionite radical anion (S2O4•−) produced by reduction of sulfur dioxide in nonaqueous solvents (Rinker and Lynn, 1968; Gardner et al., 1981; Potteau et al., 1999). If this compound is produced by photolysis of dithionite, then an aqueous electron would also be a product [Eq. (2)]. Therefore, both an oxidant (S2O4•−) and a reductant (eaq−) would be formed.
In the current study, batch experiments were conducted in solutions prepared in the laboratory to evaluate the kinetics of photochemical precipitation of selenite and selenate from water with the dithionite/UV ARP. UV irradiation is an attractive activation method for ARPs, because it is widely applied in advanced oxidation process and disinfection processes. The objective of this study is to evaluate the kinetics of Se(IV) reduction at neutral to basic pH and to discuss possible reaction mechanisms. Kinetic models of dithionite photolysis and selenium removal were developed to characterize removal and nonlinear regressions were conducted to estimate quantum yields and second-order rate constants used in the models. The result of this study will provide information to support further investigations of dithionite/UV ARP as a potential candidate for treatment of more complex selenium-contaminated water matrixes, such as surface waters, drinking waters, and wastewaters.
Materials and Methods
Reagents
All reagents were used as received. Sodium selenite (≥98%) was purchased from Sigma-Aldrich (St. Louis, MO). Sodium dithionite (powder, 88%) was purchased from Avantor Performance Materials (Center Valley, PA). Sodium selenate (anhydrous, 99.8%, metal basis), potassium hydrogen phosphate (anhydrous, 98%), and potassium dihydrogen phosphate (99%) were purchased from Alfa Aesar (Ward Hill, MA).
Experimental procedure
All irradiation experiments and related work were conducted in an anaerobic chamber (Coy Laboratory Products, Inc., Grass Lake, MI) that was filled with a gas mixture (95% nitrogen and 5% hydrogen; PRAXAIR Distribution, Inc., Byran, TX) and equipped with an analyzer for oxygen and hydrogen, fan box, and a palladium catalyst STAK-PAK (Coy Laboratory Products, Inc.) that scavenges oxygen. The anaerobic chamber was vacuumed and refilled with gas mixture and ultra-high purity nitrogen as required to maintain anaerobic conditions. The deionized water (Milli-Q; Millipore) used in all experiment was deoxygenated by sparging with ultra high purity (UHP) nitrogen for 2 h outside the chamber and then for 24 h inside the chamber. All UV irradiation experiments were carried out in 17-mL, cylindrical, UV-transparent quartz reactors (Starna Cells, Inc., Atascadero, CA) without stir. The UV light source was a Phillips TUV PL-L36V/4P UV low-pressure mercury lamp (UV-L), which emitted short wave UV radiation with a peak at 253.7 nm. The light intensity at the top of the reactor was measured with a UV digital light meter (Model No. UV 512C; General Tools, New York City, NY), which was calibrated by modified ferrioxalate actinometry (Murov et al., 1993).
Batch kinetic experiments were conducted at different pH conditions and the extent of selenium degradation was measured. The initial concentrations of target compounds, sodium dithionite, and phosphate buffer were 5, 200, and 10 mmol/L, respectively, for all experiments. The UV light intensity incident to the reactors was 1,000 μW/cm2 for all experiments. A total of 20 samples for each experiment were taken at sampling times of 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 20, 25, 30, 40, 60, 90, and 120 min for analysis of soluble selenium. The concentration of selenium in the samples was measured by an inductively coupled plasma mass spectrometer (ICP-MS).
Another batch experiment was conducted with dithionite/UV-L ARP and Se(IV) at pH 8 to study resolubilization. The initial concentrations of Se(IV), sodium dithionite, and phosphate buffer were 5, 500, and 10 mmol/L, respectively. The UV light intensity incident to the reactors was 6,000 μW/cm2. A total of nine samples were taken at sampling times of 0, 2, 4, 8, 16, 32, 64, 128, and 256 min for analysis of selenium. The concentrations of different oxidation states of selenium in the samples were measured by an atomic absorbance spectrometer (AAS).
The presence of dithionite was characterized by measuring the UV absorbance at 316 nm with a UV-Visible spectrophotometer. Before analysis, the samples were passed through a 0.2-μm cellulose nitrate membrane filter (25 mm-diameter; Whatman, Piscataway, NJ).
Analytical methods and spectroscopic characterization
An ICP-MS was used for analysis of soluble selenium (NexION 300D; PerkinElmer, Waltham, MA) (EPA method 6020A). Before analysis, the filtered samples were diluted in 1% v/v HNO3. A UV-Visible spectrophotometer (Agilent 8453; Agilent Technologies, Inc., Santa Clara, CA) was used to measure the concentration of dithionite using its absorbance at 316 nm. The concentration of dithionite was calculated using a literature value for the molar absorptivity of purified dithionite of 8,043 L/mol-cm at 316 nm (McKenna et al., 1991).
A continuous hydride generation flame atomic absorption spectrometry (HGAAS) (iCE-3500; Thermal Scientific) equipped with a VP 100 continuous flow vapor generation system (Thermal Scientific) was used to determine concentrations of different oxidation states of selenium (Se(IV) and Se(VI)) (EPA method 7742). A solution of 2.0% m/v NaBH4 in 1.0% m/v NaOH was used as reductant to reduce selenite to H2Se, which then was stripped from aqueous solution to the flame AAS for determination of the concentration of selenium. A solution of 6 mol/L HCl was used as acid reagent. All samples and standards were prepared with 10% v/v HCl. For total Se measurement, the sample was pretreated by acid digestion to reduce Se(VI) to Se(IV). The acid digestion was conducted as follows: 5 mL sample/standards and 5 mL concentrated HCl were mixed in 40-mL borosilicate glass vial. Then, the vial was loosely caped and placed in boiling water bath for 30 min. The following parameters were used for the analysis: wavelength of 196.0 nm, band pass of 0.5 nm, lamp current of 75%, background correction of D2 quadline, measurement number of 3, measurement time of 4 s, measurement delay of 60 s, pump speed of 40 rpm (which provides a sample flow rate of 9.4 mL/min, a reductant flow rate of 2.0 mL/min, and an acid reagent flow rate of 1.0 mL/min), and carrier gas (Argon) flow of 200 mL/min.
Kinetic models
A kinetic model was developed to describe dithionite photolysis and it was based on five assumptions: (1) only dithionite absorbs light and so it is the only compound being photolyzed; (2) the reactor is completely mixed; (3) photolysis is the only reaction in the system; (4) light flows in only one direction; (5) the reactor has a constant depth. The rate of dithionite photolysis is equal to the product of a quantum yield, the molar extinction coefficient, the molar concentration of reagent, and the photon flux, as shown in Equation (3).
where r1 is the rate of photolysis of dithionite at a particular point in the reactor (mol/cm3·s); φ is the quantum yield at wavelength λ (mol/Einstein); ɛ′ is the base e molar absorptivity at wavelength λ of dithionite (L/mol/cm), (note that ɛ′ = 2.303 ɛ, where ɛ is the base 10 molar absorptivity); I is the photon flux at wavelength λ at a particular point in the solution (Einstein/m2/s); and C1 is the concentration of dithionite at that point (mol/L).
Because the photon flux decreases through the reactor as dithionite absorbs light, the rate of photolysis will decrease proportionally. However, an average rate can be calculated by integrating the rate across the reactor [Eq. (4)]. The change in photon flux through the reactor can be described by the Beer-Lambert-Law and the assumption of complete mixing leads to the assumption that the concentration of dithionite remains constant throughout the reactor.
where L (cm) is the length of the light path through the solution; and I0 is the photon flux at wavelength λ entering the solution (Einstein/m2/s). The incident photon flux was calculated from the light energy flux that was measured by UV meter with the application of Planck's equation.
The model for dithionite photolysis was developed by combining the material balance equation for dithionite in a batch system with the average photolysis rate [Equation (5)]. This differential equation can be solved by integration to obtain Equation (6), which was used in nonlinear regressions (MATLAB function nlinfit) with experimental data to determine the quantum yield.
where C10 is the initial concentration of dithionite (mol/L).
An empirical second-order rate equation was chosen to describe the removal of selenium from solution, as shown in Equation (7).
where C2 is the concentration of selenium (mol/L) at specific time t (min); and k is the second-order rate constant (L/mol/min). The rate constant k was estimated by doing nonlinear regressions on selenium concentrations that were measured before the start of resolubilization. These regressions used a numerical solution of Equation (7) after combining it with Equation (6). MATLAB functions ode45 and nlinfit were used to solve the differential equation and conduct the regression, respectively.
Results and Discussion
Se(IV) removal
Results of kinetic experiments with selenite (Se(IV)) are shown in Fig. 1. Figure 1a indicates that dithionite combined with activation by UV-L was able to remove Se(IV) very rapidly during the first 15 min of the reaction time at pH 7, 8, and 9. This removal is believed due to reaction with active reducing radicals produced by dithionite photolysis. Two photolysis mechanisms have been proposed to describe this process and each produces a different reducing radical. The more conventional mechanism is that dithionite photolysis produces the same radical as does nonphotolytic degradation, that is, the sulfur dioxide radical anion. The rate of radical production by nonphotolytic reaction was estimated to be more than 50 times higher than the maximum expected rate of production of these radicals by photolysis. This calculation used a published rate constants for the non-photolytic reaction (Creutz and Sutin, 1974) and the conditions used in the experiments reported here (dithionite concentration = 2E − 4 mol/L, molar absorptivity (base e) = 8,760 L/mol·cm, irradiance = 1 mW/cm2). This means that the conventional mechanism is not likely to be the cause of the selenium removal observed. The alternative mechanism that produces the aqueous electron as the reducing species could be the correct mechanism, since processes that produce the aqueous electron have been shown to be effective in reducing selenium (Puranen et al., 2010; Duan et al., 2017).

Effect of pH on Se(IV) degradation by dithionite/UV-L ARP.
However, resolubilization occurred at later sampling times and selenium concentration was observed to increase. One hypothesis for precipitation-resolubilization is that the ARP with dithionite is rapidly reducing Se(IV) to solid elemental selenium and then the solid is resolubilized by reaction with an oxidant. The first step of this mechanism is supported by reports that formation of amorphous elemental selenium would occur from reactions of selenious acid with sodium dithionite at low pH [Eqs. (8) and (9)] (Geoffroy and Demopoulos, 2009).
The proposed mechanism for resolubilization is based on the observation that elemental selenium is not stable in the presence of UV irradiation. Compounds that could interact with UV irradiation include those formed by dithionite photolysis, which include thiosulfate (S2O32−) and sulfite species (HSO3−, and SO32−), as shown in Equation (10) (Lister and Garvie, 1959; Spencer, 1967; Burlamacchi et al., 1969; Wayman and Lem, 1970; Danehy and Zubritsky, 1974; Holman and Bennett, 1994; De Carvalho and Schwedt, 2001).
Sulfite could be photolyzed to form the sulfite radical and aqueous electron [as shown in Equation (11)].
The sulfite radical can act both oxidant and reductant (Liu et al., 2013b), but in this case, it is more likely that the sulfite radicals are oxidizing elemental selenium to form soluble selenium species. Therefore, selenium removal in the first 15 min observed in Fig. 1a is the result of formation of elemental selenium by reduction of selenite, while later resolubilization of elemental selenium is due to oxidation of elemental selenium to soluble forms by oxidants such as sulfite radicals.
The initial concentration of Se(IV) was 5 μmol/L for kinetic experiments at all pHs, but it was found that there was a large decrease in Se(IV) at pH 7 at the beginning of the experiment. This indicates that dithionite alone is able to remove Se(IV) rapidly, which is in agreement with previous studies (Geoffroy and Demopoulos, 2009; Duan et al., 2017). However, dithionite is more stable at basic conditions than that at acidic conditions (Geoffroy and Demopoulos, 2009), so there are smaller decreases in selenium concentrations at the beginning of the experiments at pH 8 and 9.
Figure 1b shows that dithionite was completely consumed within 20 min and the concentration of dithionite decreased at a similar rate for pH 7, 8, and 9. This is consistent with the soluble selenium concentration profile. The resolubilization of selenium occurred around 15 min of the reaction time because there were much less dithionite available in the solution to promote selenium precipitation.
Resolubilization study
Results of dithionite/UV-L ARP mechanism experiments with selenite (Se(IV) are shown in Fig. 2. The concentrations of Se(IV), and total Se were measured by HGAAS and the concentration of Se(VI) was calculated by subtracting the concentration of Se(IV) from total Se concentration. The concentration of Se in the solid phase was calculated by subtracting the total Se concentration from the initial Se concentration. Figure 2 shows that dithionite was completely consumed within 8 min. The concentration of total soluble Se shows a substantial decrease from the initial concentration at the first sampling time due to the reaction that forms elemental selenium and this behavior is in agreement with screening results and previous study (Geoffroy and Demopoulos, 2009; Duan et al., 2017). The total soluble Se concentration increases and the solid Se concentration decreases after the initial sampling time due to resolubilization of elemental selenium. The concentrations of Se(IV) and Se(VI) both increased after the first sampling time, with the concentration of Se(IV) showing a slight decrease at the end. However, the concentration of Se(VI) was larger than that of Se(IV) during the entire experiment, which signifies that elemental selenium was being solubilized, possibly by reaction with oxidizing radicals such as SO3•− that could be generated by photolysis of sulfite.
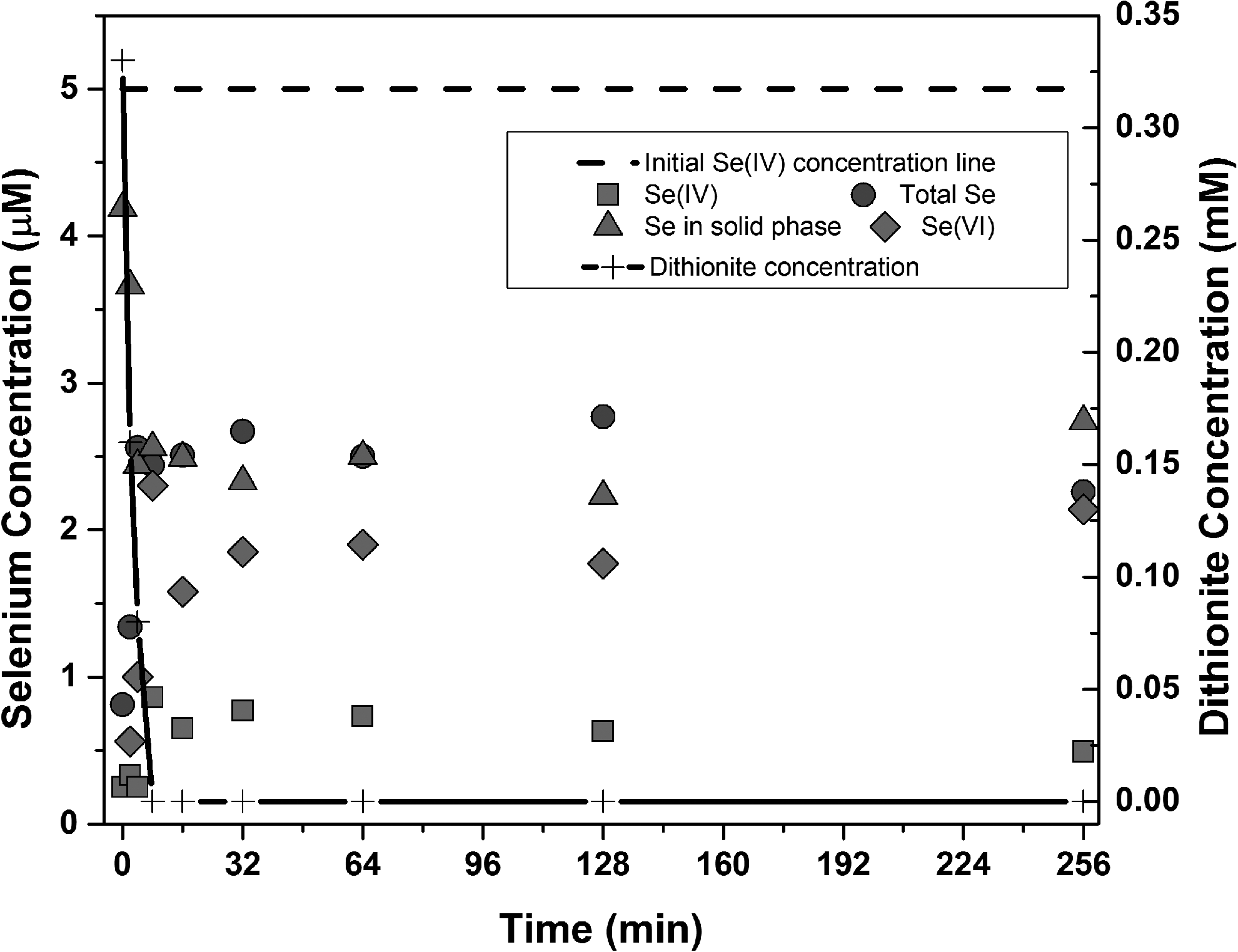
Identification of products of Se(IV) degradation by dithionite/UV-L ARP at pH 8. Initial Selenium(IV) concentration = 5 μmol/L, initial dithionite concentration as prepared = 500 μmol/L, UV light intensity = 6,000 μW/cm2.
Se(VI) removal
Results of experiments to evaluate selenate (Se(VI)) removal with the dithionite/UV-L ARP are shown in Fig. 3 as show similar results as were obtained for Se(IV). For all pH values tested, the soluble Se concentration decreased with time during the first 20 min, which was the time period when dithionite was present. Some indication of resolubilization was observed but it was much less pronounced that in Se(IV) experiments. In fact, at pH 8 soluble Se concentration continued to slowly decrease during the experiment. The rates of Se(VI) removal were slower than those for Se(IV) removal, which should be expected because Se(VI) requires more electrons to be reduced to elemental selenium. Compared to Se(IV), there were little drop in initial concentrations of Se(VI) the beginning of the experiments. This indicates that dithionite without UV irradiation was not able to effectively remove Se(VI) from water at all pH, which was in agreement with previous screening experiments (Duan et al., 2017).
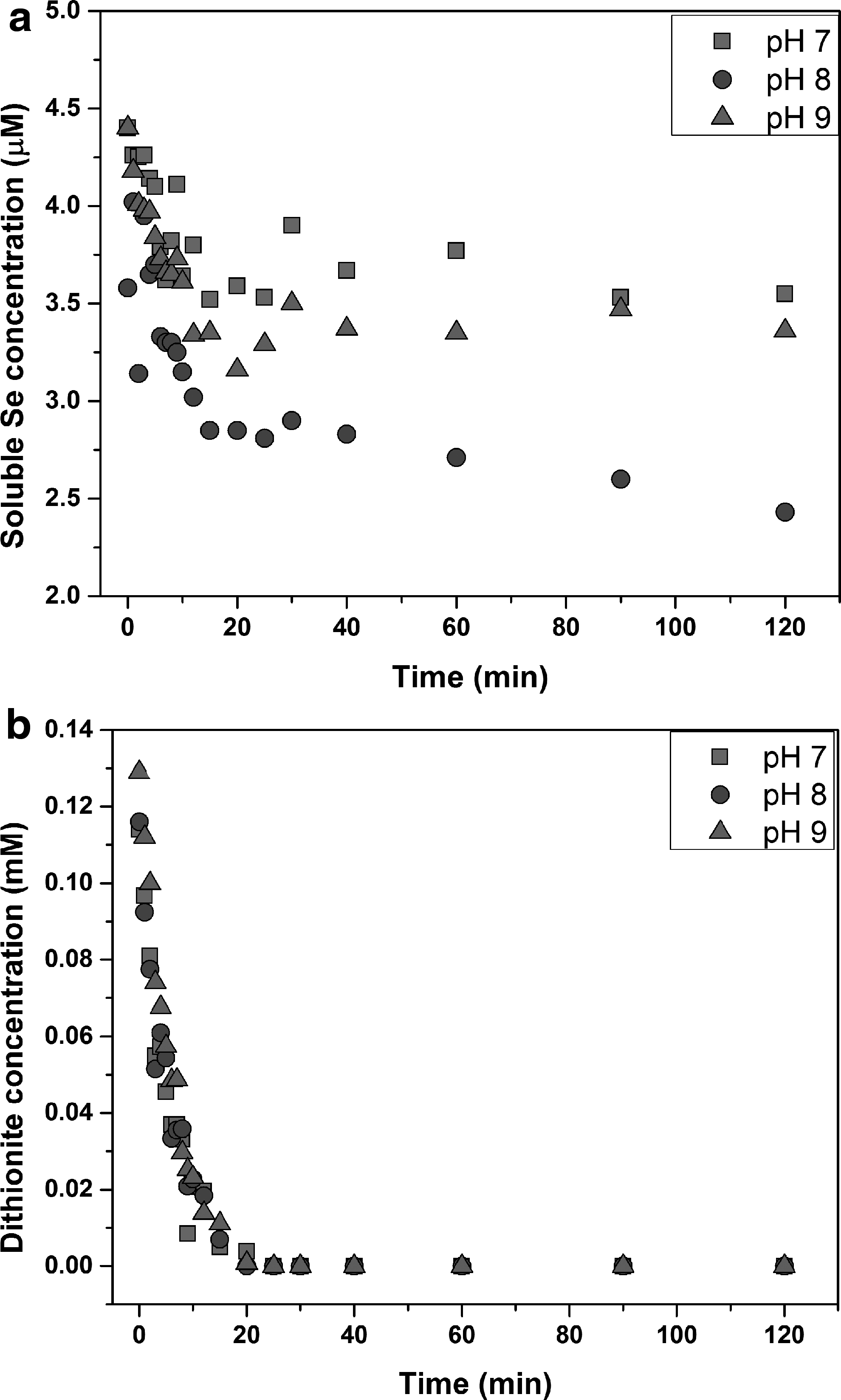
Effect of pH on Se(VI) degradation by dithionite/UV-L ARP.
Figure 3b shows the concentration profiles for dithionite. Similar results were obtained as found in experiments with Se(IV). Dithionite concentrations decreased continuously and approached zero within 20 min for pH 7, 8, and 9.
Kinetic modeling
Data of dithionite photolysis and selenium removal before resolubilization occurred were utilized to calculate the quantum yield of the photochemical reaction of dithionite and the second-order rate constant for removal of soluble selenium.
The quantum yields at pH 8 tended to be lower than those at pH 7 and 9, but the effect was minor for Se(VI) removal, as shown in Fig. 4. These quantum yields represent the overall efficiency of the process of removing selenium from solution by a series of reactions that are initiated by absorption of light by dithionite. Many factors associated with these reactions could affect the quantum yields, including formation of secondary radicals, such as sulfite radicals, which could be responsible for resolubilization of elemental selenium.
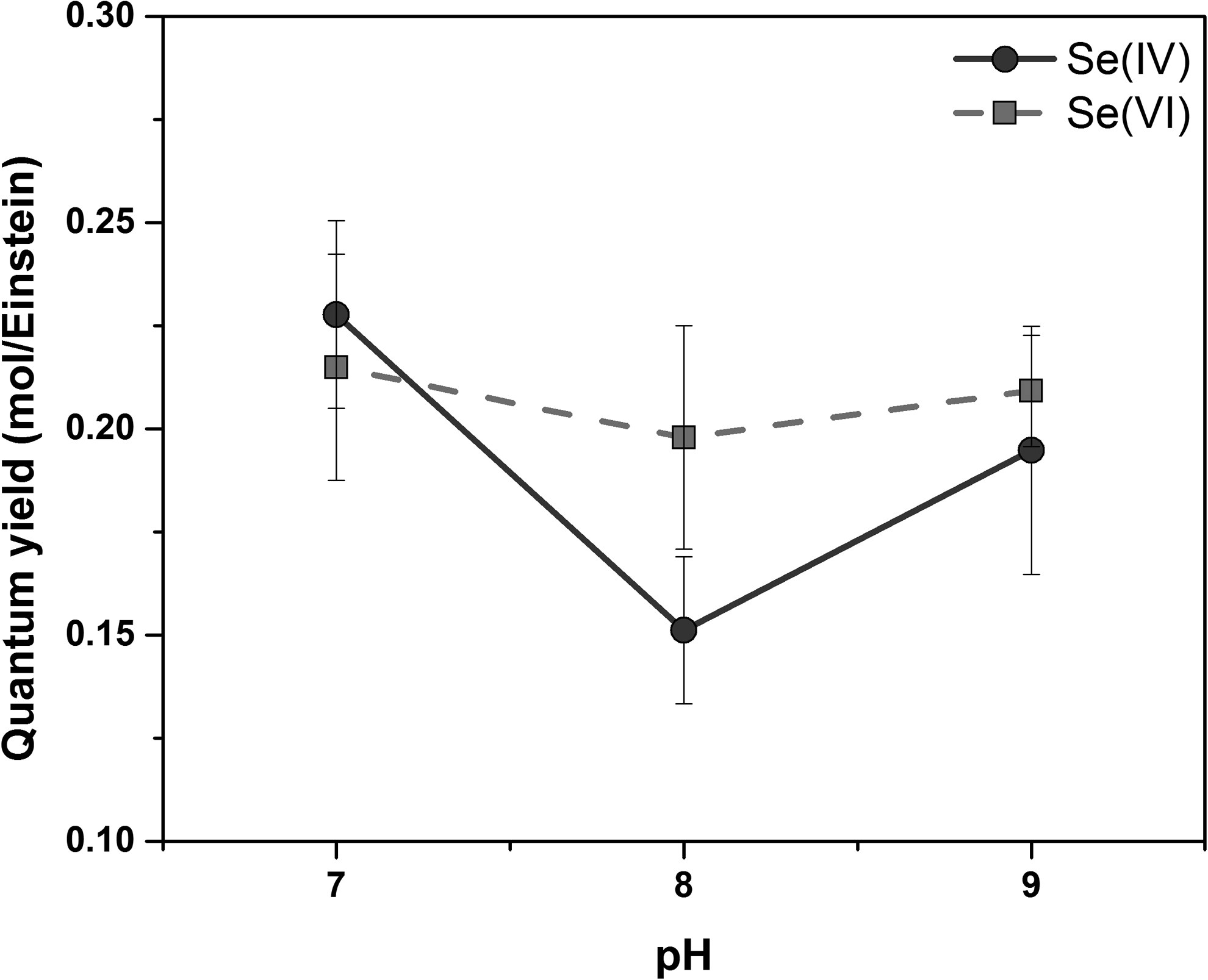
Quantum yields for dithionite photolysis at different pH. Initial dithionite concentration as prepared = 200 μmol/L, UV light intensity = 1,000 μW/cm2.
Values of second-order rate constants are shown in Fig. 5, where the error bars represent 95% confidence intervals. There was little effect of pH on the second-order rate constants for selenium removal. However, the second-order rate constant for selenate removal at pH 8 was a little larger than those at pH 7 and 9. These results are similar to the results for the quantum yields in that little effect of pH was noted, but different in the small effects observed. For selenite removal, even though the quantum yield for dithionite photolysis at pH 8 was lower, it did not affect the selenite removal kinetics. For selenate removal, there were fewer differences among the quantum yields at different pH, but faster kinetics was observed at pH 8. The real reaction scheme for removal of soluble selenium is much more complex than indicated by a simple second-order kinetic model and some of the assumptions used to derive the photolysis model may not be fully valid. Dithionite was assumed to be the only light-absorbing compound in the solution. However, selenite could also absorb light and photolyze to produce selenite radicals [Equation (12)], in the same way that sulfite photolyzes [Equation (11)] (Chawla et al., 1973; Liu et al., 2013b). Furthermore, not only the intermediates produced from dithionite photolysis, but also dithionite by itself might be able to reduce selenite to elemental selenium (Geoffroy and Demopoulos, 2009; Duan et al., 2017). The removal of selenite with the dithionite/UV-L ARP is likely due to the combined effects of these reactions.

Second-order rate constants for selenite and selenate removal with dithionite/UV-L ARP at different pH. Initial Selenium concentration = 5 μmol/L, initial dithionite concentration as prepared = 200 μmol/L, UV light intensity = 1,000 μW/cm2.
Conclusion
The dithionite/UV-L ARP is able to rapidly remove Se(IV) and Se(VI) from synthetic solutions. Although resolubilization is a challenge with applying this technology, but it can be managed by maintaining non-zero dithionite concentrations in the system and possibly by adding scavengers for sulfite radicals. The removal of Se(IV) and Se(VI) with dithionite/UV-L ARP at neutral to basic pH was rapid and was due to the formation of elemental selenium, while the resolubilization is hypothesized to be due to reactions of sulfite radicals with elemental selenium to form soluble species. The sulfite radicals could be formed by photolysis of sulfite, which is formed when dithionite decomposes. Dithionite and selenium removal data were fitted with kinetic models and they were used to determine the quantum yields and second-order rate constants. There was little effect of pH on quantum yields or second-order rate constants overall, but there was a little tendency for lower quantum yields and higher rate constants at pH 8.
Footnotes
Acknowledgments
This work was supported by the Qatar National Research Fund under its National Priorities Research Program award number NPRP 6-729-2-301. The statements made herein are solely the responsibility of the authors and do not necessarily represent the official views of the Qatar National Research Fund.
Author Disclosure Statement
No competing financial interests exist.