
Abstract
Abstract
This work aimed to explore the dynamic properties and mechanisms between natural organic matter and engineered nanomaterials (NOM–ENMs) interactions in aqueous solutions with the existence of monovalent and divalent cations. Due to the limitation of experimental method characterizing the interactions at the atomic level, the molecular dynamics simulation method was used in the study. Comparative interaction processes between NOM and TiO2 nanoparticles in the aqueous solutions with or without Ca2+ and Na+ ions were simulated. Results showed that the Temple-Northeastern-Birmingham (TNB) encircled the TiO2 nanoparticles and formed a horseshoe-style binding by two hydrogen bonds in the absence of cations. However, in the presence of cations, strong attraction between the mediating divalent cations and the negatively charged COO− groups induced the OCOO− atoms to move away from the surface, which resulted in a hinge-style binding. The less TNB fragment encircling the nTiO2 might decrease the steric and electrostatic repulsion between TiO2 nanoparticles coated by humic acid. That is, the Ca2+ might destabilize the TiO2 nanoparticles in the presence of NOM. The mediating effect of Na+ cation on NOM–ENMs interaction is insignificant, whereas the adsorbed Na+ on the surface of the nTiO2 might change the surface charge and potentially affect the environmental behavior of the nTiO2. This study facilitates to understand the fate and potential toxicity of ENMs in the aquatic environments.
Introduction
I
In natural waters, the NOMs coexist with ions. Studies have shown that humic substance can stabilize colloidal particles through steric and electrostatic repulsion in the presence of monovalent cations (Thio et al., 2011). However, some of divalent cations, such as Ca2+, Cu2+, and Pb2+, could diminish the stabilization effect of NOM (Ottofuelling et al., 2011). The destabilization effect of the Ca2+ ions on the stability of TiO2 nanoparticles was presumably caused by the electrical double layer compression and ion bridge effect between nanomaterials coated by humic acid (Zhang et al., 2009; Liu et al., 2013). In addition, it was also attributed to the complexation between Ca2+ and the humic substance. However, the microscopic mechanism of the destabilization is still unclear. Thus, a molecular thermodynamic view of NOM–ENMs interactions in aqueous solutions with the presence of monovalent and divalent is needed.
Since the interaction process between NOM and nanomaterials in the presence of cations conducts very fast and involves many atomic-level details, the current experimental observation and measurement methods are limited to adequately address the microscopic interactions and the dynamic processes at short time intervals (e.g., femtosecond). Molecular dynamic (MD) simulations provide a possible way that explores and understands the microstructure and the dynamics at the atomic level. MD has been applied to investigating the structural, dynamic, and energetic characterization of metal ions-NOM complexation and NOM aggregation in aqueous solutions (Xu et al., 2006; Kalinichev and Kirkpatrick, 2007; Ahn et al., 2008a; Iskrenova-Tchoukova et al., 2010; Kalinichev et al., 2011). The results indicate that the type of cations affects the complexation style and the form of NOM aggregation. In the presence of Na+ and Mg2+ ions (Ahn et al., 2008a), the NOM molecules aggregate spontaneously through hydrogen bonding between their functional groups, and resulting in the formation of uniformly shaped NOM clusters. However, the Ca2+ ions can strongly bind with two different NOM molecules by co-complexing the carboxylate groups (Kalinichev et al., 2011), which forms linear or branched structures. The aggregation of the NOM molecules could equilibrate much faster with more stable structures compared with other ions, such as Mg2+ or Na+, which was proved by the previous experimental observations (Brigante et al., 2007; Nebbioso and Piccolo, 2009). The MD method has proved as an excellent alternative to study the interaction process involving NOM molecules and cations. However, to the best of our knowledge, a molecular thermodynamics view of the interaction between the NOM and nanoparticle, as well as the effect of cations, are not reported yet.
To reveal the interaction mechanism and the effect of cations on the interaction process between NOM and TiO2 nanoparticles at the atomic level, we attempted to simulate the interaction process between NOM and TiO2 nanoparticles in aqueous solutions with the presence of Ca2+ and Na+ by MD simulation. By comparing the system without cations in terms of the NOM structural and functional properties and adsorption configuration, the interaction mechanism and mediation of cations during the interaction of NOM–ENMs were addressed in a nano-analysis point of view.
Simulation Details
Model system description
Two model systems (i.e., with or without ions), which contained the TiO2 nanoparticles (131 TiO2 units), NOM, and a large number of water molecules (9,957), were used to study NOM–nTiO2 interaction in aqueous solutions. The TiO2 nanoparticles were generated by removing atoms from a TiO2 crystal that were constructed using the lattice parameters for rutile (Diebold, 2003; Koparde and Cummings, 2005). A simplified neutral surface was used to present the system under pHpzc condition. The excess surface atoms (Ti and O ions) were removed to maintain a zero surface charge in the model system. Both the “Steelink” and “Temple-Northeastern-Birmingham” (TNB) models can be used to describe the atomic structures of humic acid monomers of NOM. Since the Suwannee River NOM (SRNOM) was often used as a typical representative of NOM in experiments (Roger et al., 2010), the TNB model, in which the C/O/H ratio was closer to the SRNOM (Xu et al., 2006), was selected to describe the atomic structures of NOM in the simulations. Moreover, the results of the theoretical studies using the TNB model in the large-scale MD simulations have demonstrated a good agreement with the available experimental observations (Kalinichev and Kirkpatrick, 2007; Iskrenova-Tchoukova et al., 2010). As shown in Fig. 1, the TNB model (753 Da) had three carboxylic groups, three carbonyl groups, two phenolic groups, two amine groups, and four other R–OH alcohol groups. For the pHpzc conditions (pHpzc ∼6.5) (Romero et al., 2013), it is reasonable to assume that the three carboxylic groups of NOM fragment are completely deprotonated (pKa values between 4 and 5), whereas the hydroxyl groups and amine groups are often protonated (pKa values of ∼9) (Kalinichev, 2013). The TNB molecule has a net charge of −3 e, which was compensated by adding a Ca2+ ion and a Na+ ion. As reported in a previous study, Ca2+ is the predominate cation in groundwater and river water samples, where its concentration is in the range of 4–10 mM (Keller et al., 2010). In this study, the concentrations of Ca2+ and Na+ ions were both 4.27 mM, and both were in the reasonable range. Figure 2 shows the initial conformations of the NOM–nTiO2in the absence or presence of cations. The water molecules were not displayed in the snapshot.

TNB model. First-COO−, second-COO− and third-COO− groups are illustrated respectively. TNB, Temple-Northeastern-Birmingham.
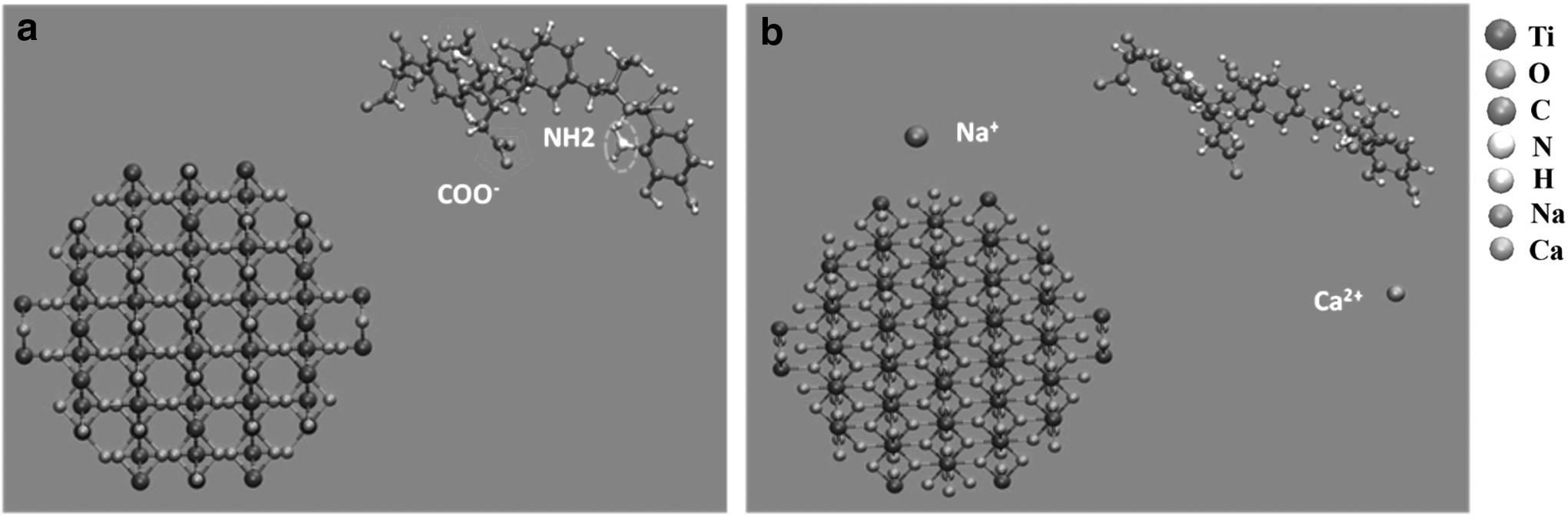
Natural organic matter and TiO2 nanoparticles model systems:
The system was hydrated by water molecules in a cubic MD simulation box with the periodic boundary conditions along the x, y, and z directions with a dimension of 73 Å. The TiO2 nanoparticles and TNB were located at the center of the model system. The nearest distance from the groups of TNB to the surface atom of the TiO2 nanoparticles was 1 nm at the initial state.
Force field and simulation methods
The Matsui and Akaogi force field is suitable for representing various titanium dioxide crystals (Matsui and Akaogi, 1991; Bandura and Kubicki, 2003; Naicker et al., 2005). Water adsorption on TiO2 nanoparticles has been well studied using the Matsui and Akaogi force field (Koparde and Cummings, 2007). In this study, the Matsui and Akaogi force field was thus applied to describe the TiO2 nanoparticle. The Van der Waals interactions were expressed by the Buckingham function. The site–site interaction could be expressed as below:
where U(r ij ) was the interaction energy between atom i and j. The Aij, ρij and Cij parameters are listed in Table 1. The interactions between the Ti atoms and the water oxygen atoms (Ti–OW) were modeled using the ab initio-derived interaction parameters reported by Bandura and Kuibicki (2003) in Table 1. The charges of Ti and O ions were +2.196 e and −1.098 e, respectively.
Interaction between the water oxygen and the Ti atom was approximated by the Buckingham function.
Předota et al. (2004) has verified that the nondissociating, rigid, nonpolarizable model is computationally efficient and yields the realistic structural results both for bulk solutions and at solid/solution interfaces down to subatomic length scales. The rigid and nonpolarizable simple point charge-extended (SPC/E) model, where partial charge was assigned to the OW (−0.8476 e) and two HW atoms (+0.4238 e), was used to describe the water molecules in this study. This force field applied the Lennard-Jones (LJ) potential to the oxygen site (Berendsen et al., 1987). The Van der Waals interactions between the oxygen atoms in rutile TiO2 nanoparticles and water molecules were treated as the O–O LJ interactions of the SPC/E water model (Předota et al., 2004). The expression of the potential was written as:
where ɛij was the depth of the potential well and σij was the finite distance at which the interparticle potential was zero. The ɛij and σij parameters are listed in Table 2. The intramolecular interactions of atoms in the NOM molecules and dissolved ions in our simulations are described by the consistent valency force field (Kitson and Hagler, 1988).
SPC/E, simple point charge-extended.
After minimizing the energy of the model system, MD simulations were conducted by using the large-scale atomic/molecular massively parallel simulator (LAMMPS) program package at a constant temperature of 300 K with an integration time step of 1 fs in the constant-temperature, constant-volume (NVT) ensemble using a Nose–Hoover thermostat (Plimpton, 1995). The cutoff distance of Rc = 12.0 Å was applied for all real-space interactions. The long-range electrostatic interactions were treated by the particle–particle particle–mesh solver, where the short-range interactions were truncated at 12 Å. Both simulations were conducted for 5.5 ns.
Results and Discussion
NOM–ENMs interaction process
First, Fig. 3 shows the variations of temperature and the total energy of the system in the presence or absence of cations. The temperature exhibited fluctuations because the velocities were rescaled at only certain intervals. However, the average temperature remained constant during the whole simulation. The total energy was also constant, except a jump due to the adsorption of TNB on the nTiO2.
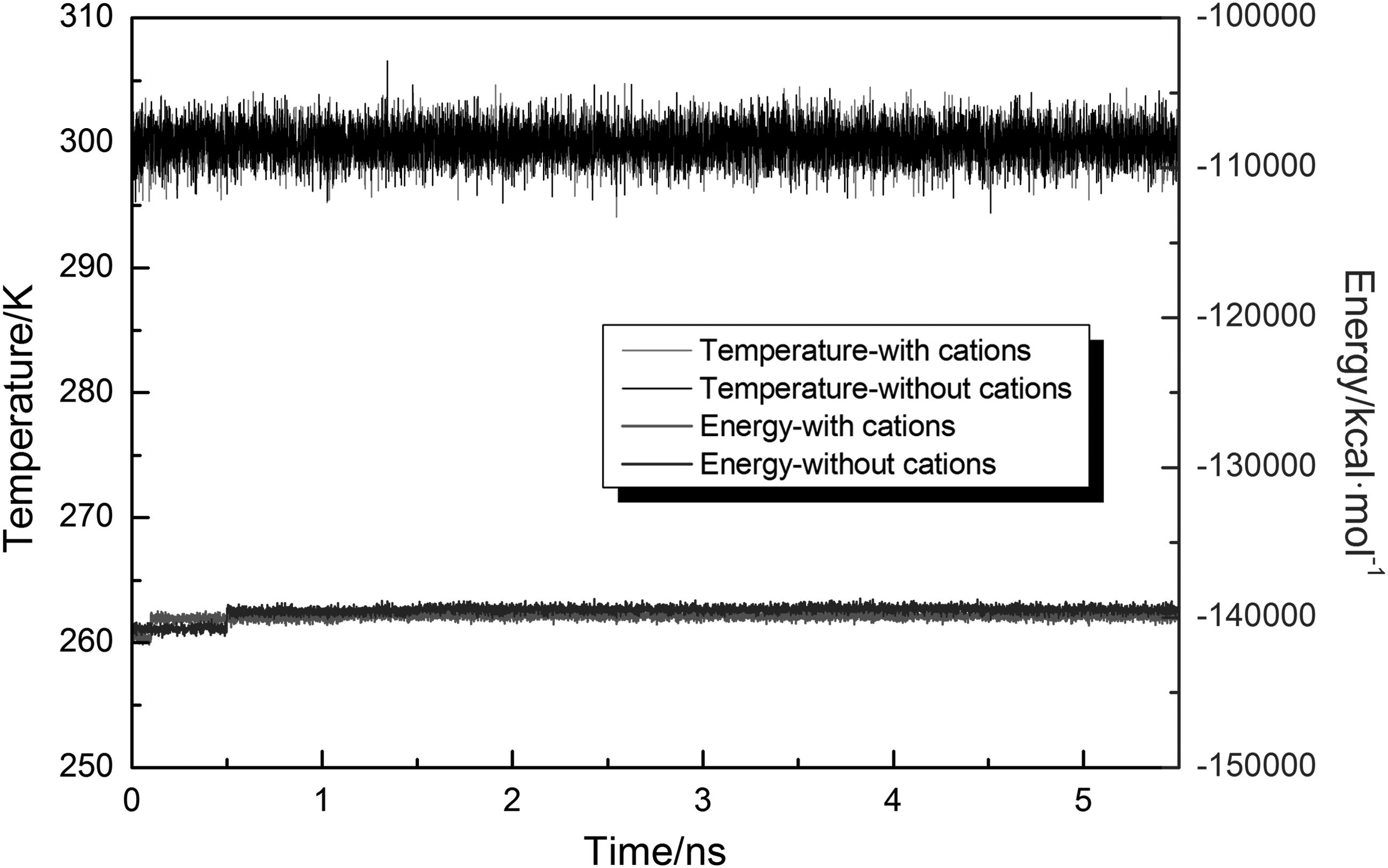
Variations of system temperature and total energy during adsorption process in presence or absence of cations.
To interpret the dynamic interaction process between NOM and TiO2 nanoparticle, the nearest neighbor (NN) distance was calculated. The NN distance was defined as the shortest distance between the atoms belonging to TNB model and TiO2 nanoparticles, respectively. The variation of the NN distance between TNB and TiO2 nanoparticles during the whole simulation period is shown in Fig. 4.

Variation of NN distance between TNB and TiO2 nanoparticles. NN, nearest neighbor.
The initial distance between TNB and TiO2 nanoparticles was 10.8 Å. In the presence of cations, the distance first increased and then decreased. At 1 ns, the distance reduced to 1.93 Å and remains stable during the rest of the simulation. The variation of the NN distance implied first that the TNB moved away from the TiO2 nanoparticles, and then moved to the vicinity of the TiO2 nanoparticle and interacted with it. In the absence of ions, the distance quickly decreased to 2.0 Å at 0.1 ns and also slightly fluctuated during the remaining simulation time. It showed that the TNB quickly approached the nanoparticles during the simulation. The results indicated that the interacting process was delayed by the cations. Furthermore, the short distance between TiO2 nanoparticles and TNB could be addressed as direct interactions between them.
To understand the interaction mechanism between NOM and TiO2 nanoparticles, the interaction conformations between the TNB molecules and the TiO2 nanoparticles in aqueous solutions were analyzed. In the presence of the cations, the terminal H atom (Hphenol) in the benzene ring of TNB was attracted by the Osurf atom in the TiO2 nanoparticles when the TNB molecule moved to the vicinity of the TiO2 nanoparticles at 1 ns, forming an hydrogen bond interaction. Evidences from ultraviolet and infrared spectroscopy also indicate that phenol groups are responsible for the attachment of humic acid to the surface (Yang et al., 2009; Erhayem and Sohn, 2014). The hydrogen bond looked like a hinge existing between the TNB molecule and the TiO2 nanoparticles. The other terminal group of TNB persistently rotated, but did not form any linking structure with bonds during the simulation. In the absence of cations, the TNB molecule moved quickly to the vicinity of the TiO2 nanoparticles within 100 ps. The hydrogen bonds were formed between the terminal H (HCOH) atoms of the TNB molecule and the Osurf atoms of the TiO2 nanoparticles at 0.2 ns. One of the terminal groups in the TNB molecules still oscillated to find the favor energy point. At 1.5 ns, one of terminal H atoms was also attracted to the Osurf atoms of the TiO2 nanoparticles, forming another hydrogen bond. Therefore, the TNB molecule was adsorbed on the surface of TiO2 nanoparticles with a horseshoe style through two hydrogen bonds until the end of the simulation run. The variation of bond lengths of these hydrogen bonds between TNB and TiO2 nanoparticles is shown in Fig. 5. All the bond lengths were smaller than 3.4 Å, which indicates the formation of hydrogen bond (Song et al., 2010; Stachowicz and Korchowiec, 2013). Due to the continuous self-regulation of the TNB, the distance of HCOH−OTiO2 was larger than 3.4 Å casually. The interaction conformation of TNB on the surface of the TiO2 nanoparticles in aqueous solutions at 5 ns is presented in Fig. 6. Due to less sites interacted and a hinge style formed between TNB and nTiO2 in the presence of cations, it can be inferred that less TNB fragment surrounded the nTiO2 in comparison to the conformations without the cations, thus the steric repulsion between TiO2 nanoparticles coated by humic acid will be decreased. That is, the cations may destabilize the TiO2 nanoparticles in the presence of NOM.

Variation of bond lengths of hydrogen bonds between TNB and TiO2 nanoparticles:

Interaction conformation of TNB on surface of the TiO2 nanoparticles in aqueous solutions at 5 ns:
As shown in Fig. 6, more COO− groups encircled the TiO2 nanoparticles in the absence of cations. To quantitatively describe the interaction between the COO− group of TNB and TiO2 nanoparticles, the NN distance was calculated. As shown in Fig. 7, all the distances between carboxyl groups and TiO2 nanoparticles changed slightly after 2 ns. In the absence of cations, the NN distances were about 6, 3, and 5 Å for the first COO−, second COO−, and third COO− groups, respectively, whereas the NN distances were 8, 13, and 5.5 Å for the first COO−, second COO−, and third COO− groups in the presence of cations. The results indicated that all the carboxyl groups surrounded the TiO2 nanoparticles in the absence of cations. However, in the presence of cations, the carboxyl groups were far from the surface of the TiO2 nanoparticles. It indicated that in the presence of cations, the surface of the nTiO2 coated by humic acid had less negative charge due to COO− groups far from the surface of nTiO2. Thus, the electrostatic repulsion between nTiO2 reduced. The less electrostatic repulsion might be another reason for the destabilization of the TiO2 nanoparticles coated by humic acid in the presence of cations.

Variation of NN distance between COO− and TiO2 nanoparticles:
In addition, the interaction energy between the nTiO2 and TNB during the adsorption process was calculated. The interaction between the nTiO2 was attractive when the interaction energy value was negative, and vice versa. As shown in Fig. 8, the interaction energy changed slightly before 2.5 ns and then dramatically decreased to −90 kcal/mol. The sudden change of the interaction energy arose from the adjustment of the interaction conformations. The interaction conformation in Fig. 8 indicates that the benzene ring near the formed hydrogen bond moved to cover above on the surface of the TiO2 nanoparticle after 2.5 ns.

Interaction energy between the nTiO2 and TNB during the adsorption process.
In general, the results suggested that the dynamic interaction process and the interaction conformation were mediated by the cations. Moreover, the mediated mechanisms for monovalent and divalent cations were investigated in the next section.
Metal cations mediating interaction between NOM–ENMs
The bulk properties of cations
In aqueous solutions, the Ca2+ and Na+ ions might interact with the water molecules and form a water shell. As shown in Fig. 9, the density profiles of the water molecules around the nanoparticles were calculated to illustrate the distribution of water layers. The peaks corresponding to water oxygen were observed at 2.35 and 2.45 Å for Na+ and Ca2+ ions, respectively. In addition, other peaks were observed at 5.05 and 4.85 Å. Similar to the water oxygen, the hydrogen profiles of Na+ and Ca2+ ions were observed at 3.05 and 5.45 Å, 3.05 and 5.35 Å, respectively. The results indicated that there were two hydration shells surrounding the Na+ and Ca2+ ions. These results were in an excellent agreement with the previous studies (Kalinichev and Kirkpatrick, 2007; Wu et al., 2012).

Water density profiles of cations.
Metal cations mediating interaction between NOM–ENMs
Variations of NN distance between the cations and TiO2 nanoparticle or TNB molecule are presented in Fig. 10. The distance between the Na+ ion and TNB quickly reduced to 6 Å. It indicated that the Na+ ion moved quickly to the vicinity of TNB at the initial simulation due to electrostatic attraction for the terminal O atoms of TNB. The distance between Na+ ion and TiO2 nanoparticles also reduced to 2.5 Å at about 1 ns. Due to the strong attraction between TNB and TiO2 nanoparticles as described in Fig. 4, it suggested that the Na+ ion moved along with the TNB to the vicinity of the TiO2 nanoparticle. Since more negative O atoms existed on the surface of TiO2 nanoparticles, the Na+ ion tended to desorb from the TNB due to the stronger attraction from the TiO2 nanoparticles and keep stable adsorption during the simulation time. The distance was kept at about 2.5 Å.

Variation of NN distances between cations and TiO2 nanoparticle, TNB molecule.
Distance between the Ca2+ ion and TNB rapidly reduced to 2.30 Å within 10 ps and remained stable during the rest of simulation time. The distance between the Ca2+ ion and TiO2 nanoparticles changed significantly at the initial stage, and it persisted at 5.2 Å after 2 ns. The variation of NN distance between Ca2+ ion and TNB indicated that the Ca2+ ion quickly moved to the vicinity of TNB and formed a stable configuration with the TNB. The distance between Ca2+ ion and TiO2 nanoparticles indicated that there is no direct interaction between them during the whole simulation.
Adsorption configuration and the variation of the NN distance between the Na+ ion and Osurf atoms of TiO2 nanoparticle are presented in Fig. 11. Water molecules within a distance of 6 Å from Na+ ion are also shown in Fig. 11. First, the inner-sphere and outer-sphere concepts were introduced to characterize the adsorption mode of ions (Wu et al., 2013). An ion in the outer-sphere mode is adsorbed at the surface through its solvation shell, whereas the inner-sphere adsorption mode is defined that the first H2O on the nonhydroxyl surface substitute water molecules from the solvation shell of ion, or no water molecules exist between the nonhydroxyl surface and ion. No water molecules existed between the Na+ ion and the TiO2 nanoparticles. It suggested that the Na+ ion was adsorbed on the surface of TiO2 nanoparticles in an inner-sphere style. This was also proved by the distance between the Na+ ion and Osurf atoms. From the discussion of the bulk properties of ion, two water layers existed around the Na+ ion. The direct attraction between the Na+ ion and the TiO2 nanoparticles suggested that the TiO2 nanoparticle had broken through the water layers around the Na+ ion and TiO2 nanoparticle. It implied that the absorbed Na+ might change the surface charge of the nTiO2, and thus affect the following environmental behavior of the TiO2 nanoparticle.

Adsorption configuration and variation of NN distance between the Na+ ions and Osurf atoms of TiO2 nanoparticle.
Adsorption configuration and the variation of the NN distance between the Ca2+ ion and the O−COO atoms of TNB are shown in Fig. 12, in which the water molecules within the distance of 6 Å from the Ca2+ ion were included. Due to the strong electrostatic attraction, the Ca2+ ion and two COO− groups formed the tetradentate coordinated contact ion pairs. It showed that the Ca2+ ion interacted with the TNB with the style of the inner sphere (Iskrenova-Tchoukova et al., 2010). According to the above discussion of the bulk properties of ions, two water layers existed around the Ca2+ ion. It suggested that the water layers around the Ca2+ ion and TNB could be broken through by the Ca2+ ion because of the direct attraction between the Ca2+ ion and the TNB.
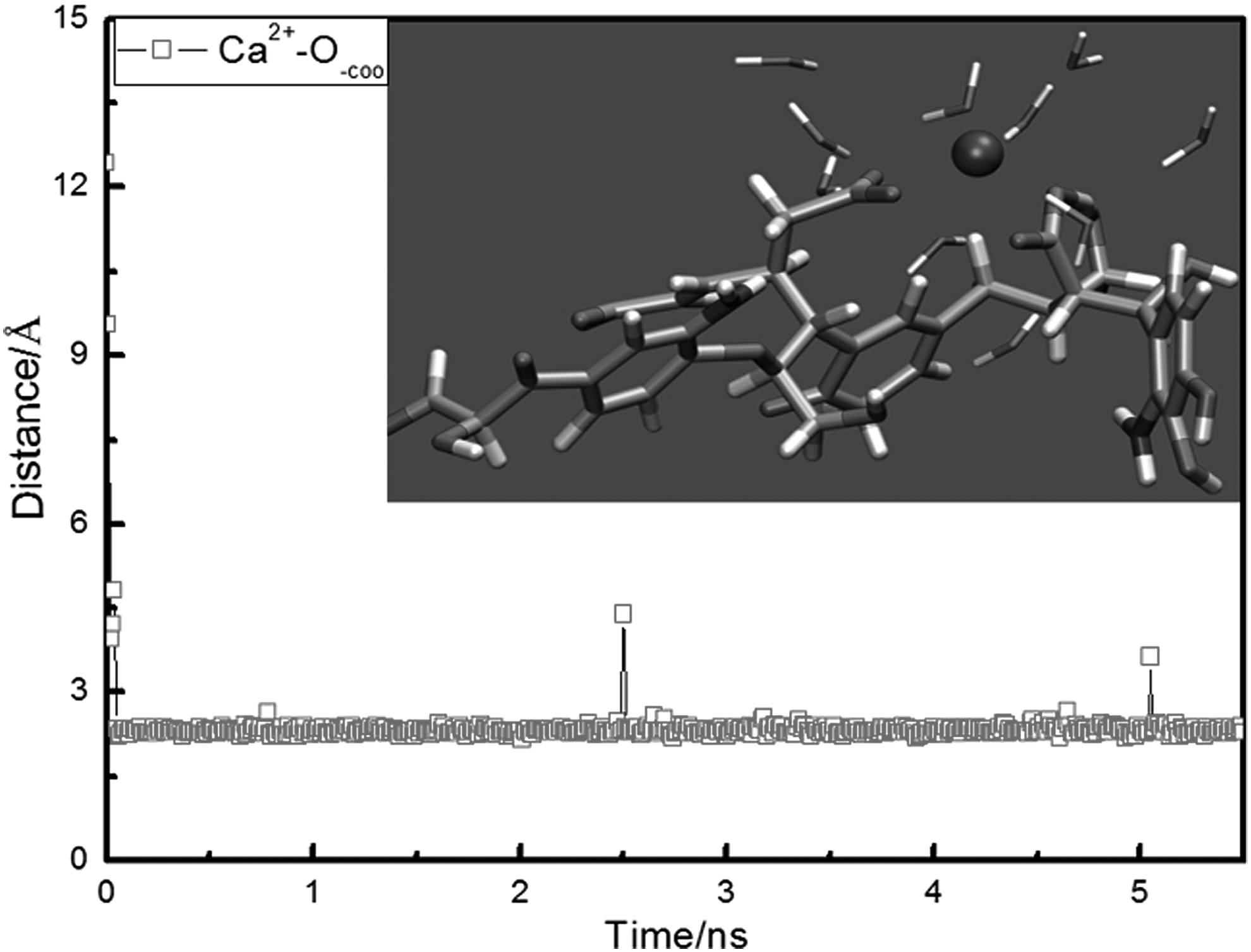
Adsorption configuration and variation of NN distance between the Ca2+ ion and O−COO atoms of TNB molecule.
In general, the Na+ ion mainly mediated with the TiO2 nanoparticles by changing the surface charge of the nTiO2 to potentially influence the following environmental behavior of the nTiO2, while the Ca2+ ion mediated with the TNB due to the strong attraction for the two COO− groups, to play an important role in the NOM–TiO2adsorption process.
Discussion on Mediating Mechanism
Based on the above results, the difference of Na+ and Ca2+ ions in mediating mechanisms during the NOM–TiO2adsorption process was discussed. The adsorption configuration in the absence of ions showed that the carboxyl groups of TNB encircled the TiO2 nanoparticles through a strong electrostatic attraction. The presence of Ca2+ decreased the adsorption affinity and changed the adsorption configuration between NOM and TiO2 nanoparticles due to a strong attraction with carboxyl groups of NOM, whereas the effect of Na+ ion is insignificant in mediating the adsorption process.
There were three structurally distinct carboxylic groups in the TNB molecular model of NOM used in our simulations, which were negatively charged. Both the Na+ and Ca2+ ions could be associated with these carboxylic groups through electrostatic attraction. The Ca2+ ion was observed to associate with two carboxylic groups and form tetradentate coordinated contact ion pairs in terms of relatively stable coordination geometries. The Ca2+ ion was coordinated with the two oxygen atoms of the carboxylic group simultaneously and stayed approximately equidistant from both of them predominately near the bisector plane orthogonal to the plane of the carboxylic group. The water molecules in the first coordination shell of Ca2+ were partially replaced by the carboxylate oxygen. The results suggested that Ca2+ ion was more effective and stable than Na+ ion in mediating the interaction between TNB and TiO2 nanoparticles. It suggested that the mediation strength was related to the charge of the cation.
Kalinichev et al. (2011) reported a similar view on the metal ion–NOM complexation. They suggested that the relative strength of the metal ion–NOM complexation could be interpreted in terms of simple electrostatic considerations, which was described by the charge/radius (z/R) ratio of the cation. Ca2+ is almost the same size as Na+ (R: Na+, 0.97 ± 0.06; Ca2+, 1.03 ± 0.05) (Marcus, 1983), but interacts more strongly with the carboxylate groups in NOM due to its greater z/R (Na+,
Although no mediation interaction between Ca2+ and the TiO2 nanoparticle was observed in our study, the mediation interaction between Na+ and the TiO2 nanoparticle was obvious. The absorbed Na+ on the TiO2 nanoparticle by an inner-sphere style might change the surface charge of the TiO2 nanoparticle. Thomson et al. (1996) also pointed out that the surface charge of the mineral can be neutralized in the presence of sufficient Na+, resulting in much more positively charged surfaces as good adsorbents for the negatively charged adsorbates. Divalent cations, in particular Ca2+, showed the capacity for effectively neutralizing negative surface charge of the mineral surface. The mediating effect between Ca2+ and TiO2 nanoparticle that was neglected in our study might attribute to the strong attraction with carboxyl groups of NOM and the less number of Ca2+ ions in the model system. Further research was expected on the mediation interaction between massive cations and TiO2 nanoparticles.
Conclusion
The MD study of the dynamic properties and the mechanisms of NOM–ENMs interactions in the existence of Na+ and Ca2+ ions in aqueous solutions provided a new quantitative insight into the dynamics and structure of NOM with TiO2 nanoparticles and the effects of metal cations on the interaction between NOM and TiO2 nanoparticles. The simulation results showed that the TNB combined with the TiO2 nanoparticles formed a horseshoe style in the absence of cations. When the cations were present, the strong attraction of the mediating Ca2+ ion to the negatively charged COO− group led to the OCOO− atoms to move away from the surface, which resulted in a hinge-style binding. The mediation strength was related to the charge of cation. The Ca2+ ion was more effective and stable than the Na+ ion in mediating the interaction between TNB and TiO2 nanoparticles. The Ca2+ ion interacted with the TNB with the style of an inner sphere. The strong electrostatic attraction between Ca2+ ion and two COO− groups formed tetradentate coordinated contact ion pairs. The Na+ adsorbed on the TiO2 nanoparticle by an inner-sphere style might change the surface charge of the TiO2 nanoparticle, thus, affecting its following environmental behavior. Our results agreed with the previous experimental findings and could substantially explain the effect of Ca2+ on the stability of aqueous nanoparticles in the presence of NOM. Also, this mechanism was able to address the complexing action of Ca2+ with NOM with respect to the fouling of the nanofiltration membranes in water purification (Chen et al., 2006; Zhang et al., 2009; Tian et al., 2013).
Footnotes
Acknowledgments
This work was financially supported by the Funds from State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2017TS03), the Creative Research Groups of China (51121062), and the National Natural Science Foundation of China (51278147).
Author Disclosure Statement
No competing financial interests exist.