
Abstract
Abstract
Heavy metal contamination of surface and ground waters from anthropogenic sources presents a significant risk to human health and the environment. Leaching of metals such as lead, cadmium, and zinc from historic mining residuals has led to extensive groundwater contamination, for example, the elevated concentrations found at the Oronogo-Duenweg Superfund site in Joplin, Missouri, United States. Excessive wastewater buildup and storage at historic mines have also caused the release of metal toxicants into river systems, as was the case in the catastrophic 2015 Gold King mine spill into the Animas River in Western Colorado. Prevention of metals contamination and the reclamation of contaminated water for human and agricultural use are compelling reasons to improve heavy metal remediation technologies. Permeable reactive concrete (PRC) is a novel, cementitious material that demonstrates substantial removal capacity for heavy metals from aqueous solutions at the bench scale. This study investigated breakthrough testing of PRC using a synthetic groundwater solution of lead, cadmium, and zinc at concentrations similar to reported values for the Oronogo-Duenweg site of ∼0.4 mg/L for lead and cadmium and 45 mg/L for zinc. Breakthrough testing elucidates removal mechanisms and reaction rates, but has never before been performed on PRC. Removal mechanisms documented in this study were precipitation of the hydroxide metals, complexation, and sorption of metals into the hydrated cement paste or metal precipitates. Demonstrated removal became more permanent over time, with total testing time around 260 days without breakthrough. Column breakthrough testing timeframes exceeded initial bench scale isotherm estimates by a factor of 20. Cost estimates for PRCs are ∼1/6th to 1/12th the cost of comparable technologies for similar site applications. PRC has been shown in this study to perform equal to, or greater than, comparable technologies and could significantly reduce remediation costs for other contamination sites.
Introduction
Motivation
H
Permeable reactive barriers
Some of the most common methods of polluted ground water remediation are pump and treat, air stripping, or passive PRBs (Mulligan et al., 2001; Hashim et al., 2011). PRB technology is an U.S. EPA preferred method of remediation because cost and efficiency are well balanced (Powell et al., 1998). PRBs are designed to passively intercept contamination plumes in groundwater with a reactive medium such as ZVI, permanganate, or activated carbon (Blowes et al., 2000; Wilkin and Puls, 2003; Henderson and Demond, 2007). PRBs are typically installed as a funnel-and-gate-system downgrade of the contaminant source to convey and capture the plume through a series of vertical walls comprising the reactive material (Trezek, 1986; Interstate Technology & Regulatory Council and Itrc, 2011). At the most basic level, PRBs remove or reduce toxicity using reduction, adsorption, and precipitation to target specific contaminants (LaGrega et al., 2001). A PRB of activated carbon was selected for remediation at the Oronogo-Duenweg site and used as the baseline material for performance comparison in this study (U. S. EPA, 1990).
Permeable reactive concrete
PRC is a novel form of PRB that uses a permeable concrete structure as the reactive medium (Holmes et al., 2017). The term PRC was first described by Holmes et al. to distinguish PRCs from standard permeable (pervious) concrete. PRC is a more porous form of permeable concrete enhanced to improve removal of heavy metals by maximizing removal sites. Furthermore, the cementitious paste is modified to enhance pollutant removal for the contaminant(s) of concern. Permeable concrete is traditionally used as a stormwater management best practice and is generally constructed using single-sized coarse aggregate, cement, and water (Kevern et al., 2008; American Concrete Institute, 2010; Holmes et al., 2016). Although most concrete is made with portland cement (PC), sustainable construction requires the use of supplementary cementitious materials (SCMs) such as fly ash. Fly ash is a byproduct of the coal combustion process and has been shown to remove heavy metals from water by adsorption or flocculation (Theis and Wirth, 1977; Roy et al., 1991; Bayat, 2002; Mingyu et al., 2011; Yuna, 2016). Permeable concretes made with PC, with or without fly ash, have been shown to purify water through physical and chemical removal mechanisms such as filtration, sorption, ion exchange, precipitation, and coprecipitation (Anderson et al., 2013; Sañudo-Fontaneda et al., 2014; Holmes et al., 2016).
Permeable concrete is typified by a porous matrix of cement and aggregate and is designed to have >15% by volume interconnected and tortuous voids (American Concrete Institute, 2010). Permeable concrete is a balance between strength with durability achieved at a lower porosity and clogging resistance achieved at much higher porosity. When applied to traditional infrastructure, such as low-volume roads, highway shoulders, and sidewalks, the permeable concrete tends toward the lower end of porosity, around 20%, to minimize raveling and improve freeze–thaw durability (Ababneh et al., 2003; Wang et al., 2014; Williamson and Isgor, 2016). However, by greatly increasing the porosity, surfaces for reactions can be enhanced to improve contaminant removal, and is one of the major distinguishing properties of PRC beyond conventional permeable concrete. A thorough examination of porosity is critical to understanding PRC and to explaining the removal mechanisms. Porosity in PRCs fall into three overlapping categories of macro-, meso-, and microscales. Each scale has a unique relationship between the pore size and localized fluid transport behavior and consequently they have different removal mechanisms.
In general, porosity in PRC describes effective porosity, or the bulk porosity, and falls between 15% and 31%, by volume. The effective porosity is the connected network of macroscale voids, or the visible holes seen between each cement-coated aggregate piece. During higher velocity flow, most of the water travels through the macropores. Principles including tortuosity, Carmen–Kozney relationships, or Darcy's law are applicable at the macroscale, as described by other researchers (Haselbach and Freeman, 2006; Montes and Haselbach, 2006). Sand and silt sized particulates are removed at the macroscale due to physical filtration caused by the tortuosity and dead end pores space (Haselbach et al., 2006; Deo et al., 2010; Kevern, 2015). Sufficiently large precipitates are also removed in a comparable way through physical filtration (Technology Transfer Division and EPA, 1980; Coleman et al., 2005; Aziz et al., 2008).
Groundwater movement is slower, and is in a flow regime nearer to Stokes flow. At Stokes flow, fluid dynamics depend mostly on viscous effects, resulting in dispersion and advection as the primary modes of physical interaction for the bulk solution within the pore space. Physicochemical properties also begin to dominate interactions between the pore solution and mesoscale cement voids as precipitation, adsorption, and ion exchange occur (Odler, 2003; Gineys et al., 2010; Kevern and Schaefer, 2013). The mesoscale cement paste contains ∼20% voids, by volume. Cement products require about a 10% less molar volume than the original crystalline components before hydration, thereby causing additional voids and shrinkage cracks to form as the cement hydration reaction progresses (Bentz, 1995). Under magnification, the mesoscale pores describe the voids visible alongside the cement phases, such as unhydrated cement grains, bleed water channels, entrapped air, and larger entrained air bubbles.
At the microscale to nanoscale, the voids in cement interact strongly with ions in solution causing chemical properties to dominate. Ion exchange, sorption, and diffusion are the primary mechanisms for mass transfer between the void and bulk solutions. Cement paste is ∼40% voids at the microscale to nanoscale, with ∼20% of the voids occurring in the calcium silicate hydrate (CSH) wherein the chemically bound water resides (Powers, 1958). At about 10 μm, cement reaction products are visually identifiable through scanning electron microscopy (SEM). Fickian diffusion controls the transport of the metal hydroxides, carbonates, complexes, and various ions into and out of the cement paste (Seveque et al., 1992; Neithalath, 2006; de Vera et al., 2015).
The chemistry of heavy metal removal in PRCs is not fully known. However, the major components of removal are documented (Knox et al., 2012; Tilson, 2013; Holmes et al., 2016). Primary removal is caused by the localized pH of 12.0 standard units (S/U) in the pore water due to the cement hydration reaction. At high pH, lead, cadmium, and zinc have decreased solubility. In addition, the ionic strength of the pore water stemming from the composition of cement reduces solubility by increasing the saturation index of the heavy metals (Technology Transfer Division and EPA, 1980; Wingenfelder et al., 2005). Cement components such as calcium, hydroxides, carbonates, sulfates, and silicates are readily available in the pore water during and after hydration. The various ions not only increase the ionic strength but also promote reactions forming insoluble products, such as calcium heavy metal double hydroxides or heavy metal carbonates (Peters and Ferg, 1987; Chen et al., 2009). Secondary removal occurs through sorption wherein metal hydroxides, formed because of high pH, favorably adsorb onto and then diffuse into the calcium-rich cement and further into the coarse aggregate, if a limestone or dolomite is included (Stipp et al., 1992; Sturchio et al., 1997; Weng and Huang, 2004; Elakneswaran et al., 2009; Calkins et al., 2010; Ernst et al., 2016; Holmes et al., 2016). Even at late ages, permeable concrete maintains a pH >9.0 S/U (Haselbach and Thomle, 2014). Although the effluent water will have a pH exceeding WHO standards for drinking water, many soils have a moderate amount of self-buffering capacity for attenuation of the high-pH water (MacNaughton et al., 2011; World Health Organization, 2012).
Column testing
Column testing, also known as breakthrough, fixed-bed, or packed-column testing, is a method of measuring contaminant removal rates under expected operating conditions (Powell et al., 1998; Tillman Jr. et al., 2005). The complex process of removal in PRC necessitates column testing to identify the dominant mechanism(s) because each mechanism greatly influences future engineering design options. Fouling by clogging and saturation of the reactive surface areas are two of the primary design considerations. Columns are typically made of tubing between 10 and 100 cm in length and sufficient width to overcome wall effects due to interactions between the media and the tubing (Powell et al., 1998). The column is then filled with reactive media so the influent solution can pass through the material (by gravity or pump). Aliquots of effluent are collected to analytically measure contaminant concentrations over time. Temperature, influent chemistry, and flow rate are typically held constant to simplify description of reaction kinetics. Column testing for the removal of heavy metals from solution has been extensively used for PRB media designs (Reed et al., 1994; Dwivedi et al., 2008; Purnomo et al., 2008; Turner et al., 2008; Brooks et al., 2010; Li and Benson, 2010; Mandadi, 2012). Small scale breakthrough testing using 50 × 100 mm permeable concrete samples and rapid flow has been performed by Knox et al. (2012). However, large scale testing at slower, groundwater flow rates has not been investigated. This study investigated the breakthrough response of synthetic groundwater (SG) contaminated with lead, cadmium, and zinc and passed through basic PRC made with PC or class C fly ash (CA).
Experimental Methods
Design considerations
The breakthrough system was designed to be representative of field conditions for the Oronogo-Duenweg Superfund site in Missouri, United States. The design flow rate was 16 mL/min and corresponds to the contaminated aquifer flow of ∼1.5 m/day, typical for the region (Czarnecki et al., 2009). Design concentrations for lead, cadmium, and zinc were 0.39, 0.36, and 45.44 mg/L, respectively. The values were selected to represent extreme conditions of an idealized worst-case contaminant loading scenario (U.S. EPA, 1990). The pH of the influent, heavy metal laden, SG was held constant at 6.5 S/U.
Breakthrough column designs for PRC or permeable concretes have not previously been established. Therefore, the column diameter, length, and material were evaluated to minimize adverse system-based effects, such as wall effects, and produce results representative of the PRC material. Wall effects in the lateral and longitudinal directions have a significant influence on the performance of breakthrough (Delgado, 2006; Li and Benson, 2010; Pugliese and Poulsen, 2014; Popa et al., 2015). Wall effects due to placement of permeable concrete are known and require a minimum diameter of 100 mm (Kevern, 2015). The fluid dynamic wall effects of packed columns also recommend a 7:1 ratio of diameter-to-grain size for laminar flow (Arbuckle and Ho, 1990). Pipe with 150 mm diameter and 10 mm wall thickness made from acrylonitrile butadiene styrene (ABS) plastic was selected to provide sufficient cross-sectional area to limit wall effects while providing adequate strength to support the fresh PRC during casting. Internal cracking of the concrete from movement before full strength is achieved could modify the structure by artificially increasing the available reactive sites, preventing the formation of a representative specimen. ABS was chosen for the wall material because a wall thickness of 10 mm provided sufficient rigidity to minimize internal cracking from movement during system setup. Black ABS was used to minimize biological influences by restricting the available light.
Mixture proportions and final design
Two mixture proportions were tested using Type I/II ordinary PC (labeled PC) conforming to ASTM C150/C150 M with a Blaine fineness of 373 m2/kg and a Bogue composition of 55% C3S, 17% C2S, 8% C3A, and 10% C4AF (ASTM C150, 2016; Cao and Kevern, 2015). The control mixture contained no SCM, whereas the second mixture had 25% replacement PC with CA (labeled CA) adhering to ASTM C618 (2010). Limestone aggregate conforming to ASTM C33 was used with a nominal diameter of 8 mm and 0.9% water absorption (ASTM C33, 2003; ASTM C127, 2015). PRC was mixed and placed according to ASTM C192 and C305 (ASTM C305, 2014; ASTM C192, 2016). Mixture proportions are shown in Table 1.
PC, portland cement; CA, class C fly ash.
Approximately 30 kg of PRC was used per column. Compaction was performed every 150 mm using three blows of a standard marshal hammer (ASTM D6926, 2016). Columns were stored in an environmental chamber (23°C, 100% relative humidity) for 2 weeks then removed and sealed with endcaps. Angle pipes were used to direct flow between the top and bottom of columns, ball valves were used for deairing of columns and prevention of airlocks, and a needle valve was used at the end column to regulate outflow. A constant head of 1 m was used as the driving pressure and was used in the Carmen–Kozney equation [Eq. (1)] to estimate final porosity (Trussell and Chang, 1999).
where ΔP is the pressure head loss for the column, L is the length of the column, g is acceleration due to gravity, vs is the empty bed velocity, D is the diameter of the particle, and Φe is the effective porosity. Figure 1 is a schematic of the column layout with initially six columns in series for each mixture.
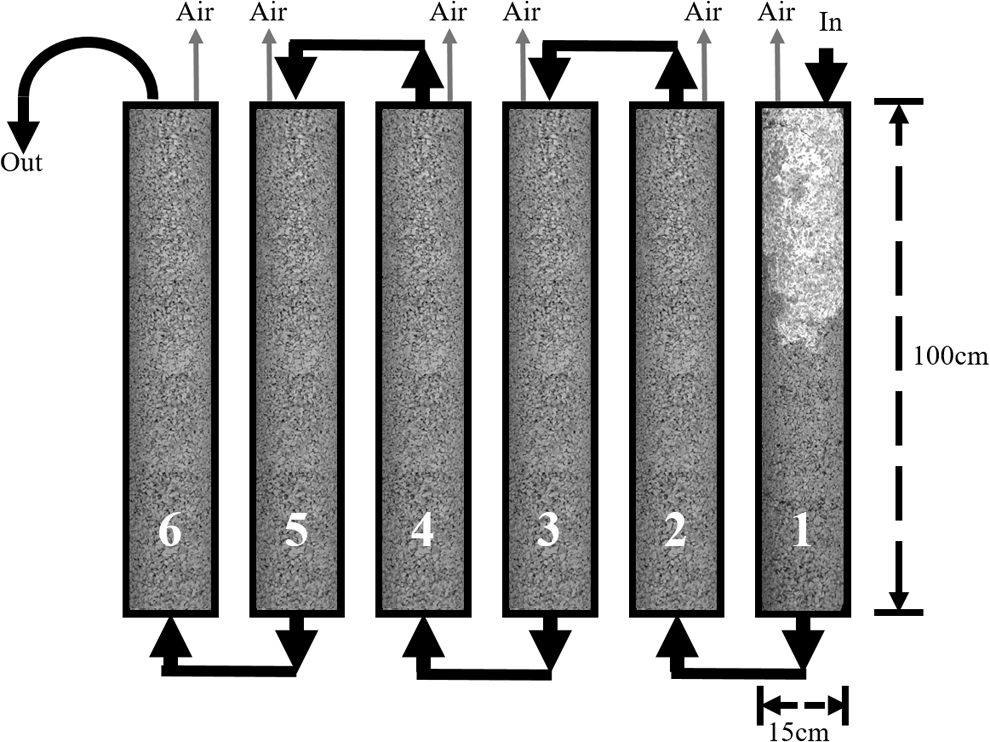
Schematic of breakthrough column design configuration.
Previous batch-sorption testing produced isotherms were used to calculate the approximate time until breakthrough (Holmes et al., 2016). The calculation estimated one column worth of reactive material would be required per day, given the mass of each column, design concentration, and the total column length. Isotherm calculations estimated that saturation of one column would require ∼1–2 days, which, in retrospect, resulted in a substantial overdesign of the system. Aliquots were taken over time after influent solution progressed through the columns (Fig. 2). After 117 days, breakthrough had not occurred between the first and second columns, nor were metals detectable between any of the final four columns. The six-column setup was then modified by moving the needle valve from column 6 to column 2, thereby reducing the system to two columns. The design modification continued functioning until testing was halted at 266 days.
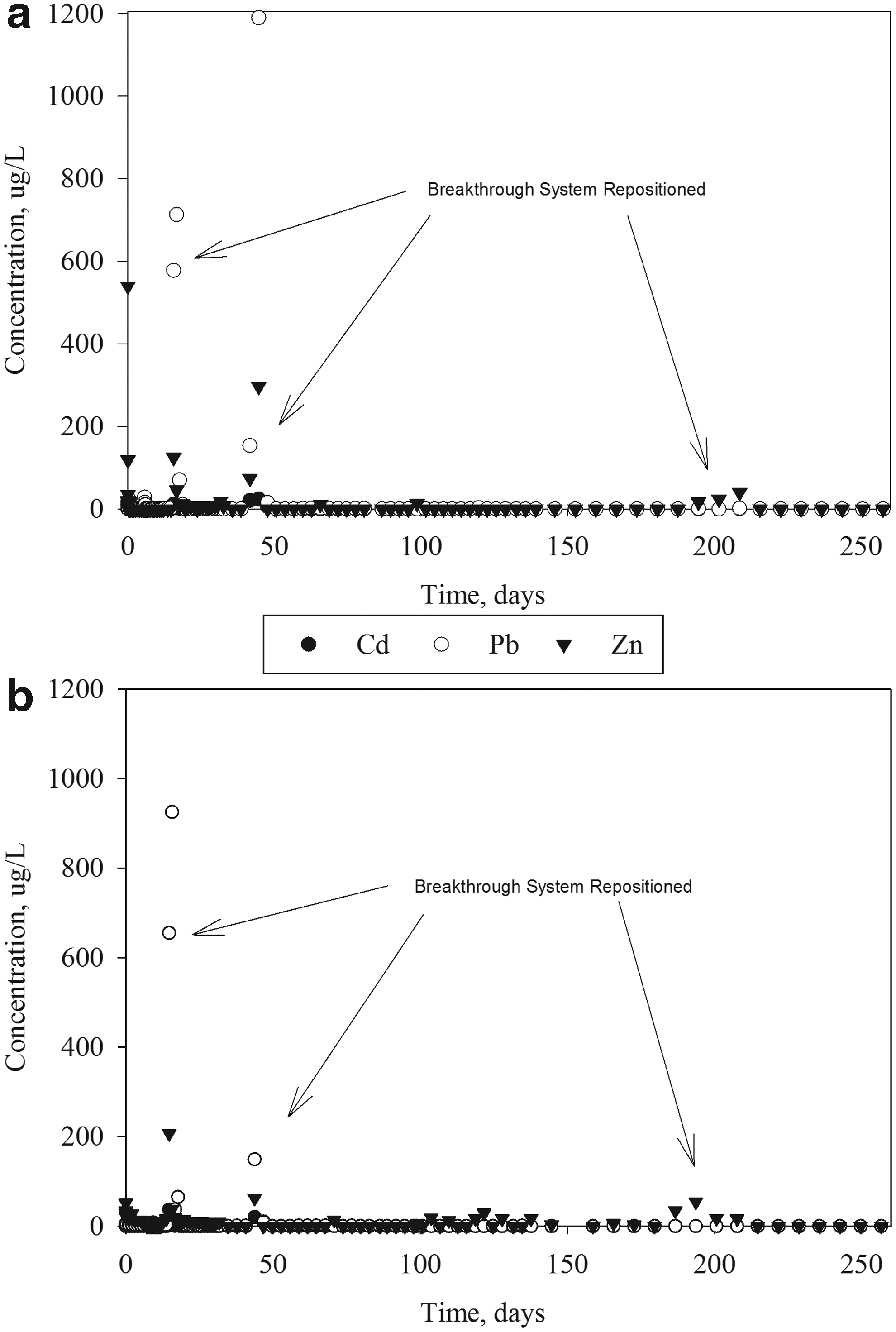
Concentration of metals in aliquots from each mixture PC
Column setup and sampling protocol
Concentrated stock solution of 50:1 of the design concentration was produced using ASTM D1193 deionized water and ACS grade lead chloride, zinc chloride, and cadmium chloride (ASTM D1193, 2011). The SG was produced by diluting the concentrated solution with deionized water and then deairing the solution by vacuum pump for 12 h. The deairing process also prevented airlocks and encouraged dissolution of localized air pockets in the concrete matrix to maximize contact area (Mackenzie et al., 1999). Initially, the columns were filled simultaneously by pouring SG into the fill ports at the base of the system (Fig. 1) to push the air out as in ASTM D2434 and to prevent airlock. SG was used instead of deionized water to prevent excessive leaching of the calcium hydroxide and provide a qualitative indication of initial reaction kinetics. The first fill of columns was performed in <5 min with all air valves remaining open until columns were completely saturated. Once filled, the needle valve was opened and flow was permitted at the design flow rate of 16 mL/min. Aliquots were taken rapidly at first with samples collected at 5, 10, 20, 30, and 45 min (Fig. 2). Then samples were taken every hour until 12 h had elapsed and then every 12 h until the end of testing. Conductivity, temperature, and pH were measured for each sample from the first day and then once a day. Aliquots were then acidified with 1 mL of nitric acid and tested per EPA method 200.8 using inductively coupled plasma mass spectrometry (ICP-MS).
Results and Discussion
Breakthrough column testing
Removal greatly exceeded initial calculations and the experiment was halted at 266 days when breakthrough had not yet occurred between the first and second columns. Results of metal concentration for tested aliquots sampled from both column series (PC and CA) are show in Fig. 2. The total treated water volume was 4,170 L for PC and 4,400 L for CA, translating to ∼920 pore volumes based on design porosity. Flow rate began near the design flow rate at 16 mL/min with a linear decrease overtime at a rate of 0.04 mL/day and was on average 11.4 mL/min. The pH followed a similar decreasing trend starting at about 12.5 for PC and CA, and ending at around at 12.1 for PC or 11.8 for CA. A lower pH in the fly ash mixture was expected as the pozzolanic reaction in fly ash consumes calcium hydroxide when forming CSH, thereby reducing available hydroxides. At the end of breakthrough testing, the flow rate was no longer entirely limited by the needle valve as system fouling by precipitates at the top of the first column had reduced permeability to approximately the design flow.
Initial removal was rapid, with all metal concentrations reaching below WHO drinking water quality standards (Cd: 5 μg/L, Pb: 15 μg/L, Zn: 5,000 μg/L) within the first 10 min of flow. The concentration of metals remained below ICP-MS detection limits for the duration of the test, except for the three noted spikes caused by the repositioning of the entire breakthrough setup. The columns and piping were originally installed on a mobile trolley cart allowing for the entire system to be moved when necessary. At 16, 44, and 200 days, the trolley cart was repositioned and the vibrations of the columns caused corresponding peaks of metal concentrations, as shown in Fig. 2. Since the concentration of the peaks was higher than the stock solution, the peaks are likely due to precipitates mobilized during transport becoming solubilized in the nitric acid used for sample preservation. Lead or zinc is the most likely to mobilize within the column due to flexing of the column during transport since the metals initially form low density, poorly ordered precipitate structures (Holmes et al., 2017). Lead concentrations alone exceeded water quality standards at the peak values and only during the early stages of testing. After 200 days, the metal concentration spikes were much lower, indicating increased physical stability of precipitates with larger floc size and lower mobility. The large flocs are visibly evident as the white material shown in Fig. 1 at the top half of column 1.
At 110 days, water samples were taken from the end of each column to locate the depth of metal penetration. Nondetectable levels of metals were measured for all six columns. Since the saturation front had not reached the end of the first column and plug flow was occurring, the last four columns were removed to reduce the lifespan of the breakthrough system. The first two columns continued testing until the experiment was halted at 266 days at which time the metals front still had not penetrated the first column.
Post-testing forensics of columns
After breakthrough testing was stopped, the columns were freely drained for 24 h. Samples of each column's drained solution were tested for residual metals, and metal concentrations were measured to be below detectible limits for all columns. Column casings were then cut in two to expose the inner concrete and measure the contaminant front, as depicted in Fig. 1 by the white discoloration in the influent column. Precipitates in PC had a visible penetration depth of ∼33 cm and in CA they had a penetration depth of ∼40 cm. Both mixtures had similar precipitation patterns with some preferential paths visually identified by the distinctly white color of hydroxide or carbonate precipitates. The precipitates appeared to have filled the pores evenly across the surface of the column for the metals penetrated portion. The concrete was noticeably friable within the first 10 cm of the column, which is attributed to a loss of cement paste either by metal displacement or by diffusion, or both. Concrete strength increased in the direction of flow down the column, with the initial 5 cm easily hand excavatable, and the end of the column achieving about 8,000 kPa compressive strength. The second column showed no signs of precipitates, and exhibited no such strength loss. Fouling by the precipitates filling the macro- and mesopore spaces was observed in the first 10 cm with no observable closing of pore space beyond the initial zone. The reduction in initial pore space appears to correspond to the linear decrease in flow observed during breakthrough testing.
Since breakthrough did not occur, kinetics of removal could not be directly determined through traditional analytical methods from solution concentrations. In lieu of kinetics, effectiveness of the column was measured according to depth of metal penetration within the first column. The profile of metal distribution from the top of column 1 was used as a quantitative estimate for overall removal capacity. Acid digestion of subsamples was performed in triplicate down the length of the column in 15 cm increments. Samples were removed from the middle of the column at each 15 cm cross-section to avoid edge effects. Reference samples were collected from column 6, where no precipitates were visible, to account for the initial metal loading from the first fill. Representative samples of 5.0 g were taken from each 15-cm subsection, acidified in 250 mL of concentrated hydrochloric acid, filtered with 250 mL of additional deionized rinse water, and dried. The subsequent solutions were tested by ICP-MS for metal concentrations. The acid digestion procedure was designed to mimic metal recovery in carbonate systems and resembles EPA method 3050b for heavy metal testing of soils (U.S. EPA, 1996). A mass balance of total metals in the SG solution minus the retained metals within the concrete was performed and accounted for >85% of all metals. The average dose for each metal versus depth from the top of the column is shown in Fig. 3a as lead, 3b as cadmium, and 3c as zinc. A summary of the regression values is presented in Table 2. PC and CA showed a similar exponential trend for all metals with R2 values of 0.89 or greater as is to be expected of plug-flow systems. The lower regression and lower slope for CA shown in Fig. 3a and c are attributed to the saturation of removal sites within the first 30 cm of the column. Precipitation of the metal ions is dependent on pH and calcium concentration. It is well known that fly ash's pozzolonic reaction is a lime-consuming reaction, resulting in a lower concentration of calcium ions and reduced pH (Taylor, 1997). Therefore, although not directly measured, pore pH and calcium concentration in the CA mixture were lower than those in the control PC mixture, resulting in an increased overall depth of penetration as compared with the control.
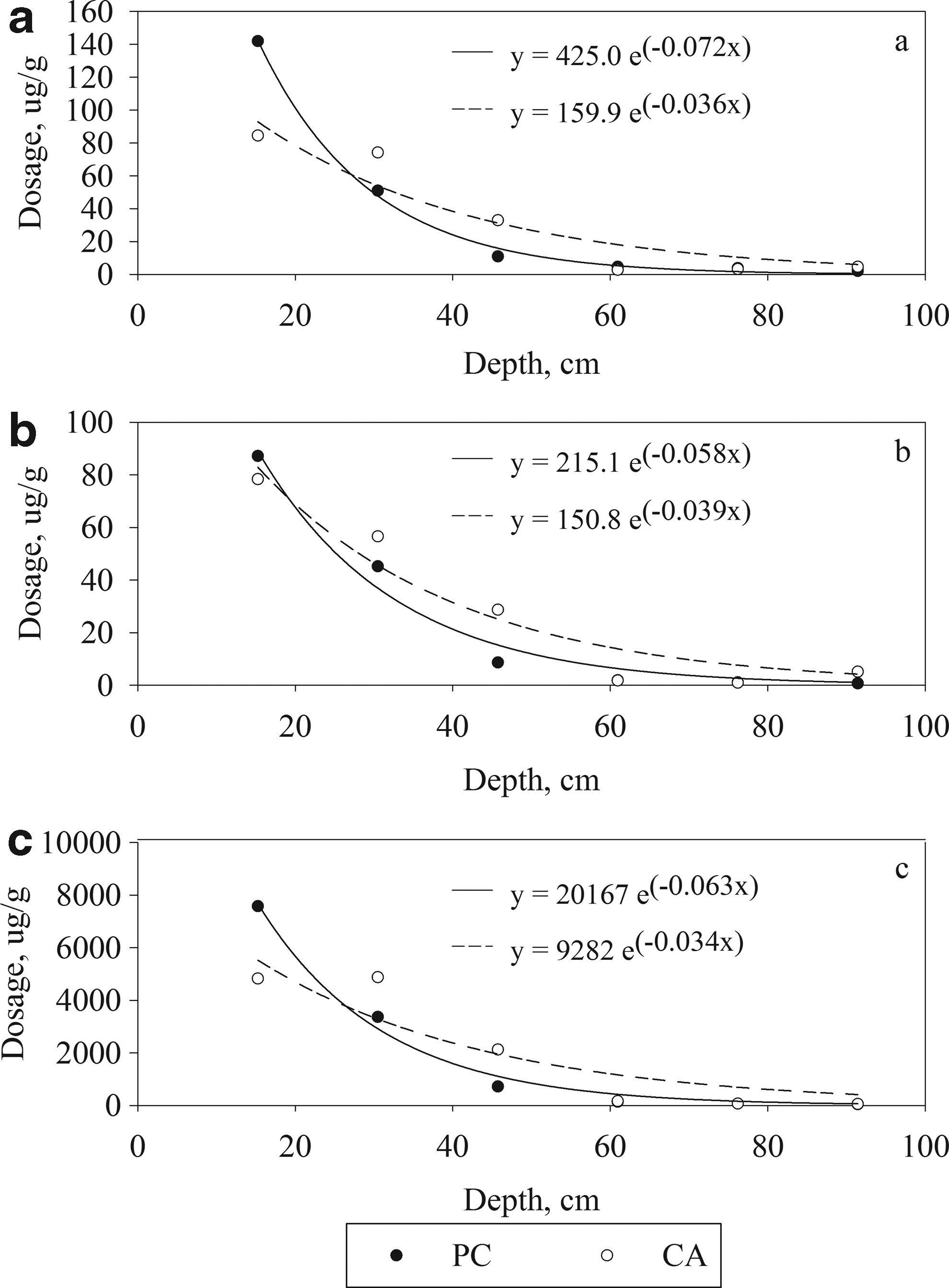
Acid-mobilized metals with increasing depth into the first column: lead
Scanning electron microscopy
Precipitation is the first and most visible step in a complex progression of removal mechanisms of metals by PRC. To elucidate the other mechanisms, such as sorption and diffusion, SEM and energy dispersive X-ray spectroscopy (EDX) were performed on subsamples from the topmost portion of the column. Two representative areas of concrete under SEM are shown in Figs. 4 and 5. EDX was performed in conjunction with the SEM images to identify and evaluate the distribution of metals.

Scanning electron microscopy backscatter image of precipitates found on the surface of cement in open pore space:

Scanning electron microscopy backscatter image of precipitates
Figure 4 shows the external surface structure of a macropore coated in metal precipitates. The structure of the precipitates shown in Fig. 4a is highly porous at the microscale, with a webbed network of low-density spheroids mainly formed from zinc as shown in Fig. 4b. The precipitate spheroids were initially self-nucleating around zinc. As the precipitates gained mass, they settled within the pore space and formed larger agglomerations. Localized lead hotspots were evenly distributed throughout the structure as shown in Fig. 4c. Lead removal occurred at preferential sorption sites, but no correlation with other ions could be found from the SEM analysis performed. Cadmium was not visible by EDX, which is most likely due to a uniform distribution below detection limits. Alternatively, the initial zinc concentration was much higher than the cadmium concentration, which may have caused blinding of the cadmium signal.
Figure 5 is a representative sample of aggregate coated with cement taken from the top of the column. The piece was fractured to allow for visual examination of the cement paste coating versus aggregate. The image shows the fracture surface. The brighter cement paste is highly concentrated with zinc. Most of the surface calcium has been displaced by zinc as seen in the contrast between Fig. 5b and c. Zinc is known to interact strongly with amorphous CSH gel, causing the formation of zincates, zinc silicates, and other thermodynamically favorable zinc–CSH structures. Zincate reactions have been shown to destabilize the CSH and change the solubility of the gel (Tommaseo and Kersten, 2002). The leaching of CSH is a slow process and has been shown to relate to the diffusion and interlayer sorption of the zinc (Tommaseo and Kersten, 2002). The destabilized CSH was evident by the friable nature of the PRC structure in the upper most zone. The interfacial transition zone (ITZ) is a relatively high porosity layer that occurs between aggregate and the hydrated cementitious paste due to particle packing effects (Oh and Jang, 2004; Zuquan et al., 2007). The ITZ contains a much higher content of calcium hydroxide (Portlandite) formed later in the hydration process. As evident by the gap between the limestone and cement paste and high concentration of zinc at the surface of the cement paste shown in Fig. 5a, the calcium hydroxide originally contained in the ITZ was leached by the SG and contributed to maintaining a high pH further down the column.
Calcium in the limestone was resistant to zinc diffusion. However, lead fully penetrated the limestone aggregate. Trace amounts of cadmium were also found in conjunction with lead. Lead removal was opportunistic, because lead has the smallest ionic radius of the three metals tested and calcium. Lead more readily was adsorbed and diffused into the cement and limestone, but by doing so, lead most likely modified the bonding structure during displacement of the calcium ions, allowing cadmium ingress. The particular limestone used was highly crystalline with a high amount of calcite, which is a well-known adsorbent material for lead, cadmium, and zinc (Davis et al., 1987; Papadopoulos and Rowell, 1988; Sturchio et al., 1997; van der Weijden et al., 1997; Godelitsas et al., 2003; Merrikhpour and JalaLi, 2012). Primary sorption occurs at the edges of the crystal structure with inner-sphere complexation promoting diffusion of the sorbed metal to the interior of the crystal (Elzinga and Reeder, 2002; Li et al., 2012). Multilayered deposition of metal carbonates and calcite could have also occurred in the spheroids, thereby providing continuous renewal of reactive sites (Zachara et al., 1988; Fernández-González, 1999). Also, outer or inner sphere sorption and heterogeneous substitutions of the metals onto or within the calcite crystal warp the crystal structure to form less symmetrical edge zones, further contributing to the sorption removal capacity of the crystals by producing alternate growth planes (Heberling et al., 2014). Similar arguments could be made for the calcium-rich cement or limestone surfaces because portlandite or hydrocalumite, both found readily in concrete, have also been shown to sorb heavy metals in a variety of mechanisms and phases (Shively et al., 1986; Ziegler et al., 2001; Chrysochoou and Dermatas, 2006; Chen et al., 2009).
Effective macroporosity was back calculated using Equation (1) and was ∼6% at the top of column 1. The precipitates in the meso- and micropores were sufficiently physically close together to cause possible ion rejection throughout the lifespan of the PRC (Song et al., 2014). In addition, as the macropores fouled, a similar phenomenon likely occurred to electrostatically “pad” the pore space and encourage larger precipitates to flow to significantly greater depths than inert particles. Typical fouling associated with physical removal in permeable concrete is within 10–15 cm of the surface (Deo et al., 2010). Depth of penetration in the breakthrough column is more than double what had been previously reported, suggesting that electrostatic forces play a significant role in the deposition and mobility of the precipitates. Similar padding has been observed in the dissolution and fouling dynamics of limestone caprock (Ellis et al., 2013).
Diffusion of the metal ions occurs in any one of three phase boundaries: liquid–solid, solid–solid, and liquid–liquid. Diffusion in the breakthrough column system describes the physical dispersion, or Brownian motion, and chemical concentration gradient, or Fick's Law with Nernst–Planck modification, and has a significant role in the ion transport and removal processes. Breakthrough testing was unable to capture the exact diffusivity constants for every boundary layer, but general relationships can be derived from the physical penetration and precipitate analysis. Based on Fig. 3, >96% of the metals penetrated ∼60 cm before the recoverable concentration approached 0. Assuming 266 days of continuous flow, a depth of metal penetration of ∼60 cm for both mixtures and a square relationship of effective liquid diffusivity depth over time for two-dimensional diffusion, diffusivity of the bulk media is ∼7.8 × 10−5 cm/s, which is comparable to previous studies on diffusion of the metals in the bulk porewater for fixed beds (Bažant and Najjar, 1972; Purnomo et al., 2008). Cement paste is highly porous with general salt diffusion into the matrix occurring primarily along the liquid–solid boundary at a rate of ∼10−9 cm/s (Bostick et al., 1999; Jensen et al., 1999; Zhou et al., 2014). Calcium leaching occurs at approximately the same rate of 1.0–2.2 × 10−9 cm/s (Marchand et al., 2001). Based on the leaching rate, after 266 days, the metals would have overcome the full depth of the estimated cement thickness (2 mm) from liquid–solid diffusion alone. Inspection by EDX of the SEM images from Fig. 4 supports this conclusion, as the cement has been completely penetrated by the zinc, and some lead and cadmium have penetrated the limestone aggregate. Upon physical examination, the precipitate penetration is directly related to strength loss in the cement paste associated with metal diffusion into, and hydroxide leaching out of, the cement. However, the depth of degradation is larger than the concentration gradient would suggest if sorption and precipitation are nearly spontaneous processes. The depth of degradation also indicates that additional forces, compounding diffusion, are likely. One contribution is possibly due to the crystallographic changes already discussed. Both dissolution and fixation of ions are expedited by the crystallographic stresses associated with the larger metal ions warping the calcite bonds and modifying surface charge densities to potentially cause localized sinks for sorption similar to pitting found on ZVI used for arsenic removal (Lackovic et al., 2000). Also, the high molecular stress zones could promote additional diffusion by creating additional reactive surface area with the formation of holes or gaps from surface sheering induced on the calcium-based minerals by the smaller ionic radius metals such as lead or cadmium. Another contribution could be at the macroscopic level wherein precipitates accumulate due to physical filtration. The locally increased liquid–solid metal concentration gradient caused by contact between precipitate and concrete could increase diffusion and promote cement degradation deeper in the column, earlier than the bulk pore water concentration gradient would suggest.
Economics of applications and effects of scale
Cost estimates are critical to any scale-up implementation of PRC as an innovative technology and provide a point of comparison between different PRB materials. Breakthrough results typically provide a baseline for estimating the material cost to remediate a site. Although breakthrough of PRC was not achieved, forensic analysis can provide an estimate for the remaining material available for contaminant removal and is based on total depth of penetration. Since the column would only fail after removal had reached some saturation point along the length of the column, two depths of penetration were used to compare estimates for the remaining capacity of the columns. Depth 1, breakthrough was about to happen at the termination of the experiment at 266 days (full column depth, 100 cm). Depth 2, breakthrough would only occur when the effluent concentrations based on Fig. 3 would have exceeded the WHO recommendations for drinking water of 5 μg/L of cadmium, 15 μg/L of lead, and 5,000 μg/L of zinc (60 cm). Option 1 had an inherent factor of safety built in because the aliquots at the end of testing were measured below detectible limits of metals. Option 2 is also conservative as the concentrations measured required liberation by a concentrated acid and were based on micrograms per gram (dose), rather than micrograms per liter (concentration). Average dosage of metals removed was measured by integrating the measured data points in Fig. 3 and dividing by the chosen depth of penetration for either option. Cost estimates were calculated by dividing the total mass of each metal to be treated by the average dosage of metal removed per gram of concrete (dose), then multiplied by the cost of PRC, estimated as ∼$100 per m3. Cost estimates are provided in Table 3 based on current market values.
N/A, not available; PRC, permeable reactive concrete.
Estimated remediation costs for the Oronogo-Duenweg site's groundwater are $60–90 million (U.S. EPA, 1990). Previous publications have performed cost analysis of bench scale testing by comparing isotherm results from activated carbon with the PRC system (Holmes et al., 2017). Preliminary estimates from the previous article show a range of removal for the Oronogo-Duenweg site from $3 trillion for lead removal by the control PRC mixture to $424 million for zinc removal. However, PRC isotherm calculations did not fully capture the diverse reactions and compounding removal mechanisms observed in this breakthrough study and over-estimated costs by orders of magnitude. New cost estimates for metal removal by PRC are from $1.2 to $1.7 million for option one and from $90 to $830 thousand for option two.
Ultimately, results must be compared with the leading sorbent utilized in industry—activated carbon. Since activated carbon comes in many varieties, lead removal isotherms used for calculations were drawn from a low-cost source, and zinc isotherms from a commercial source (Issabayeva et al., 2006; Kouakou et al., 2013). Kouakou et al. developed zinc removal isotherms using commercial grade activated carbon with pure zinc solutions and Issabayeva used low-cost palm shell-based activated carbon with pure lead solutions. The isotherms were then used to estimate the mass of activated carbon needed to remediate the Oronogo-Duenweg groundwater. The total cost for materials is between $5 million for lead and $28 million for zinc and is based on rough estimates from current market values (Holmes et al., 2017). Cost estimates for activated carbon have some limitations as market variations and sources greatly affect effectiveness of the activated carbon as well as cost. Assuming equivalent installation costs, PRC is a minimum of 5 times lower in cost than an activated carbon system for option 1, and 10 times lower for option 2. The estimates described are intended to provide an order-of-magnitude reference frame to compare activated carbon with PRC.
The comparison was based on pure systems of granular activated carbon with one solute, and isotherm values did not consider competitive ion effects. Improvement beyond previous batch reactor testing is mostly attributed to the gravitationally forced flow promoting full contact with most of the available reactive sites in the concrete. Batch reactors, although well mixed, do not have sufficiently high enough velocity to overcome the tortuosity of concrete pores, thereby limiting the overall contact area for removal. Batch reactors were tested for only 72 h, whereas breakthrough was performed for almost 7,000 h, which allowed for slower processes such as diffusion and precipitate stabilization to occur as evidenced by the resistance at later ages to the rerelease of metals when the column was disturbed.
Summary
Remediation of contaminated water is of vital importance for the sustainable supply of clean water for human consumption, agricultural use, and environmental release. Breakthrough testing is a method of measuring removal capacity and identifying removal mechanisms. Breakthrough was performed on PRC to elucidate the effectiveness of PRC in removing lead, cadmium, and zinc from aqueous solution. PRC was shown to be highly effective, surpassing initial predictions by orders of magnitude. Removal of lead, cadmium, and zinc by PRC is a complex series of processes with interconnected and interrelated subsystems. Precipitation and diffusion are the two primary mechanisms of removal. After 266 days of testing, breakthrough of the columns with mixtures of either PC or CA did not occur. Post-testing forensics revealed the depth of precipitates and provided approximations for depth of removal. The removal capacity of the columns significantly exceeded initial calculations based on batch testing isotherms, and appeared to be physically limited by precipitate fouling and chemically by the rate of diffusion. Initial removal kinetics were rapid and occurred within the first 10 min of system flow, providing removal at 100%, unless the system was otherwise physically disturbed. Physical removal of precipitates played a significant role in overall removal and is related to the pore size and tortuosity of the system. Precipitates after 266 days reached a depth of 30 cm and diffusion had a depth of 60 cm. Precipitates formed rapidly and were self-nucleating to form spheres and eventually networks of sorption sites. Fouling of the precipitates was significant and a major concern and likely to limit future design. Sorption and diffusion of the metals on and within the cement paste, precipitates, and pore water played an equally important role as precipitation. If precipitates were not the limiting factor inhibiting flow and the diffusion front was the primary source of breakthrough, the first column would have likely lasted another 6–8 months before signs of breakthrough could have been expected. Since the pore size of the selected mixture was relatively small (∼8 mm), subsequent mixtures could easily be produced with larger pore spaces.
Future work includes field testing with full-scale implementation of the PRC-based PRB. Additional testing on the concrete matrix to analyze the diffusivity in the cement or other concrete constituents, depth of precipitate penetration in the pore space, and dependence of flow velocity on removal is also needed. Correspondingly, a model needs to be developed using the results of the additional tests and known chemical parameters to estimate the sensitivity of the PRC system with regard to each removal mechanism.
Footnotes
Acknowledgments
Funding has been provided through the National Science Foundation Grant number CBET-1439378. This study is also supported by the University of Missouri Intellectual Property Fast Track Initiative. ICP-MS testing was performed by PACE Analytics.
Author Disclosure Statement
No competing financial interests exist.