
Abstract
Abstract
This study demonstrates that silver (Ag+) impregnated graphene oxide (GO) reduces anion and natural organic matter (NOM) competition for bromide (Br−) adsorption sites compared with Ag+ impregnated powdered activated carbon (PAC). We impregnated two GO (Tour and Modifier Hummers [MH] method) and one PAC with silver ions. Batch studies were conducted to assess Br− removal in model waters with Br−, chloride (Cl−), bicarbonate (HCO3−), and/or NOM and natural surface waters. In buffered ultrapure water, Tour-Ag, MH-Ag, and PAC-Ag all removed >85% of Br−, while sorbents without Ag+ removed <3% of Br−. In all water matrices, Tour-Ag removed >75% of Br−, MH-Ag removed >50%, and PAC-Ag removed >30%, highlighting that GO-Ag is more effective at removing Br− from water than PAC-Ag (p < 0.05). Scanning electron microscopy and energy dispersive X-ray spectroscopy analysis show that Br− is evenly dispersed on the surface of GO-Ag, indicating possible attachment to oxygen groups and silver on the GO surface. A leaching test of GO-Ag in buffered water showed that ∼20% of Ag+ loaded onto GO leaches into solution, of which only 1–3% remains when Br− is spiked into solution, indicating possible complexation and precipitation as AgBr. GO-Ag and PAC-Ag were introduced separately in combination with alum during coagulation and flocculation operations. Both MH-Ag and Tour-Ag showed high removal of Br−, demonstrating that GO-Ag could supplement current technologies used in water treatment facilities when Br− removal is needed.
Introduction
B
SWTPs typically use chemical oxidation such as chlorination to achieve disinfection before distributing to consumers because of its low costs and broad-spectrum biocidal potency. However, oxidants can form disinfection by-products (DBPs). Bromide can react with ozone (O3) or hypochlorous acid (HOCl) to form hypobromous acid (HOBr), which reacts with organic matter to form Br-DBPs (e.g., brominated trihalomethanes or haloacetic acids) (Kampioti and Stephanou, 2002; McTigue et al., 2014; Zhai et al., 2014; Winid, 2015). Br-DBPs are more cyto- and genotoxic than their chlorinated analogs (Plewa et al., 2008; Richardson et al., 2008; Yang et al., 2014). As water regulation becomes increasingly stringent, reducing and controlling the formation of emerging Br-DBPs is becoming more important, raising the requirement for novel technologies to effectively remove Br− before disinfection in water treatment facilities (Richardson and Postigo, 2017).
Bromide in surface waters can be removed using granulated activated carbon (GAC) (Frommer and Dalven, 2000) or powdered activated carbon (PAC) (Chen et al., 2016) impregnated with silver ions. Silver ions adsorbed onto the carbon surface can form insoluble precipitates with Br− (Ag+ + Br− → AgBr(s), Ksp = 5.2 × 10−13). However, Br− removal by silver impregnated activated carbon in complex aqueous matrices may be challenging because of competing components and low intraparticle diffusion rates. For example, the molar ratio of chloride (Cl−) to Br− in seawater and many drinking waters is between 300:1 and 1,000:1, although competing precipitation reactions with chloride (Ag+ + Cl− → AgCl(s), Ksp = 2.8 × 10−10) are less favorable than Br− (Davis et al., 1998; Mullaney et al., 2009; Katz et al., 2011). There are other ions that may compete with Br− for Ag+ on the carbon surface such as iodide (I−). In addition, natural organic matter (NOM) can block activated carbon pores or occupy sites that contain silver, likely reducing the ability of activated carbon to remove Br− from natural waters proficiently (Li et al., 2003; Chen et al., 2016).
Graphene oxide (GO), a sp2-bonded two-dimensional (2D) carbonaceous material with a sheet-like structure, can overcome such practical limitations by accommodating competing ions, NOM, and Br− concurrently and decreasing the time required for intraparticle diffusion (Apul et al., 2013; Ersan et al., 2016). Theoretically, GO has a high surface area like other carbonaceous materials, which allows for a high number of sorption sites. When in an aqueous suspension, however, the surface area of GO is slightly diminished due to aggregation. In addition, the high oxygen content of GO provides an abundance of functional groups that can be used for the impregnation of GO with silver ions. The GO functional groups have been previously used to synthesize a variety of graphene-based sorbents for cationic contaminant removal (Zhao et al., 2011; Perreault et al., 2015). We hypothesize that the 2D open nature of GO, compared with the intrapore surface area of activated carbon, will improve Br− removal when impregnated with Ag+ ions.
The goal of this study was to evaluate the potential of silver impregnated GO nanomaterials (GO-Ag) as a new sorbent material for removing Br− from surface water. The objectives of our study were to (1) determine the ability of attaching Ag+ onto functionalized GO surfaces, (2) evaluate the ability of silver impregnated GO-Ag to precipitate Br− from aqueous media, (3) compare Br− removal efficiency of GO-Ag versus PAC-Ag, (4) investigate the impact of surface water chemistry on Br− removal by GO-Ag, and (5) understand the mechanism for Br− removal by GO-Ag.
Materials and Methods
Solution preparation
Br− and Cl− stock solutions (20 mg/L) were prepared by dissolving sodium bromide (EM Science; CAS# 7647-15-6, >99% purity) or sodium chloride (Sigma Aldrich; CAS# 7647-14-5, >99.5% purity) in ultrapure water (18.2 MΩ cm; Thermo Fisher Barnstead GenPure xCAD Plus Water Purification System, Art no. 50136170). Because Br− adheres to glass, all stock solutions were prepared in plastic bottles to avoid adhesion losses. The buffered water solution (1 mM NaHCO3, pH ∼8) was prepared by dissolving sodium bicarbonate (Amresco; CAS# 144-55-8) in ultrapure water.
NOM isolate was purchased from International Humic Substances Society (CAT # 2R101N, Suwannee River NOM RO Isolation). The NOM stock solution (50 mg NOM/L) was prepared in ultrapure water. The SUVA254 value of the NOM isolate solution was 4.19 L/mg-m. The natural river water was sampled from the Colorado River at the Central Arizona Project (CAP) canal at the City of Scottsdale Water Campus (Average dissolved organic carbon [DOC] of 3.0 mg/L). The CAP water characteristics are summarized in Supplementary Table S1.
Carbonaceous adsorbent synthesis and characterization
Graphite used to synthesize GO was purchased from Bay Carbon (CAS# 7782-42-5, >99.5% purity). Two different GO materials were produced from the Bay Carbon graphite using the Modifier Hummers (MH) and the Tour (Tour) chemical oxidation protocols (Tung et al., 2008; Marcano et al., 2010). GO synthesis details can be found in the Supplementary Data. As-purchased PAC (Norit 20B M-1789) was oxidized in concentrated nitric acid at ∼90°C for 1 h to produce oxidized PAC. The complete oxidation procedure is described in the Supplementary Data.
Silver nitrate (Sigma Aldrich; CAS# 7761-88-8) was used to impregnate GOs and PAC. Silver impregnation of the different carbonaceous materials was done by dispersing 200 mg of oxidized PAC, MH, or Tour in 0.5 M AgNO3 for 2 days and then collecting the material by centrifugation and vacuum drying. Complete material preparation procedure is described in the Supplementary Data.
The carbonaceous nature of the different materials and the abundance of defects introduced by the oxidation procedure were characterized by Raman spectroscopy (full spectra in Supplementary Fig. S1). The silver content of the carbonaceous material before and after silver impregnation was determined by acidifying samples in 2% HNO3 for 24 h and quantifying total silver with inductively coupled plasma mass spectroscopy (ICP-MS; Thermo Scientific X Series II). Some silver ions may be impregnated into pores of the carbonaceous materials and may not be dissolved in 2% HNO3. There was not enough sample size to run a microwave digestion, which would have been the alternative method for silver ion quantification. The size and morphology of the carbonaceous material and silver precipitates before and after the experiments were conducted by scanning electron microscopy (SEM). Energy dispersive X-ray spectroscopy (EDAX) was coupled with SEM to identify elements in the SEM micrographs. SEM/EDAX was used to characterize the Ag+ adsorbate distribution on the carbon surface. The samples were filtered through 0.2-μm nylon syringe filters (Thermo Scientific F2500-2) to remove excess organics and carbon sorbents. The filters were air-dried, and the carbon sorbents trapped in the filters were imaged with SEM/EDAX (FE-SEM, Amray 1910). The specific surface area (SSA) of the materials was quantified by nitrogen gas adsorption at 77 K with a physio-sorption analyzer (Micromeritics TriStar II 3020). The Brunauer-Emmett-Teller (BET) equation was used to calculate surface areas from adsorption isotherms.
Bromide removal experiments
Bromide removal experiments were conducted in 125 mL plastic vials that were shaken using an in-house end-over-end rotational mixer (45 rpm). Bottle-point experiments used a 4-h contact time, which represented the hydraulic residence time for PAC treatment at water treatment plants (Westerhoff et al., 2005). Four water samples that were spiked with 200 μg/L Br− were used: (1) 1 mM NaHCO3 buffered ultrapure water, (2) 1 mM NaHCO3 buffered ultrapure water spiked with 20 mg/L chloride, (3) 1 mM NaHCO3 buffered ultrapure water spiked with 20 mg/L chloride and 10 mg/L NOM, and (4) natural CAP surface water. Each carbon adsorbent was added as a powder to a concentration of 25 mg/L. Details are shown in Supplementary Table S2. To simulate water treatment processes, bromide removal experiments using CAP water as the background matrix were conducted in 2-L jar testers (Phipps and Bird). All acrylic jars were filled with 1 L of source water and initially mixed for 6 min at 200 rpm, simulating coagulation (i.e., rapid mixing). During the rapid mixing step, 28 mg/L of alum (provided by the Scottsdale Water Campus) and each carbon adsorbent were added as a powder to a concentration of 25 mg/L. The mixing speed was later decreased to 25 rpm for 30 min, simulating flocculation (i.e., slow mixing). Mixing was ceased and the flocs settled for 1 h (i.e., sedimentation). No additives were used during the slow mixing and sedimentation steps. After sedimentation, an aliquot for analysis was withdrawn from the middle of the jar using a 50-mL plastic syringe. Careful consideration was taken to obtain the aliquot without upsetting the sediment.
Silver leaching experiments
Silver leaching experiments were conducted in 125 mL plastic vials that were shaken using an in-house end-over-end rotational mixer (45 rpm) to determine the amount of Ag+ leached into solution during mixing. Each carbon adsorbent was added to a concentration of 25 mg/L in four different water matrices: (1) 1 mM NaHCO3, (2) 1 mM NaHCO3 with 200 μg/L Br−, (3) Natural CAP water, and (4) Natural CAP water with 200 μg/L Br−. The vials were shaken for 4 h.
Measurements of dissolved species
UV254 (UV-Vis Spectroscopy; Horiba Scientific Aqualog) and DOC (SEC-DOC; Shimadzu ASI-V) were measured on filtered (0.2-μm nylon) samples. Filtered samples were analyzed for Br− and Cl− using ion chromatography (Thermo Scientific Dionex ICS-5000) to measure anions remaining in solution (IonPac™ AS18 Column; 30 mM KOH Eluent, 25 μL injection volume) following the EPA 300.1 method. Select experiments were also analyzed for Br− in solution by acidifying filtered solutions in 2% HNO3 for 24 h and quantifying total Br− using ICP-MS (Thermo Scientific X Series II).
Results
Characterization of the silver impregnated carbon sorbents
Table 1 summarizes characterization data for the PAC and GO adsorbents before and after silver impregnation and shows how their physicochemical properties are altered by the silver-adsorbent interaction. Raman spectroscopy detected G and D bands, which are characteristic of oxidized carbon materials (Supplementary Fig. S1). The G band is related to the sp2-bonded carbon lattice, and the D band is related to carbon structure disorder, which may be caused by structural defects, the introduction of new functional groups, or decreasing crystallite size (Ferrari, 2007; Ferrari and Basko, 2013). The D:G ratios of the oxidized PAC, MH, and Tour were 1.023, 0.882, and 0.834, respectively, indicating high disorder in the oxidized carbon structure (Table 1). All three carbonaceous materials had high oxygen content, with Tour having the highest oxygen content (C:O ratio of 1.787) and oxidized PAC having the lowest oxygen content (C:O ratio of 7.310) (Table 1). The differences in C:O ratios result from different oxidation conditions present during material synthesis, which is known to influence carbon material functionalization (Dreyer et al., 2014). The SSA of the three materials was 716 m2/g for oxidized PAC, 9.38 m2/g for Tour, and 0.55 m2/g for MH (Table 1). The SSA of the GO materials was noticeably lower than the theoretical value of 2632 m2/g for graphene. This lower value for GO has been observed multiple times for different GO materials and can be attributed to the tight restacking and aggregation of GO sheets (Guo et al., 2014; Perreault et al., 2015).
Images are in the Supplementary Data.
XPS, X-ray photoelectron spectroscopy; BDL, below instrument detection limit; BET, Brunauer-Emmett-Teller; PAC, powdered activated carbon; MH, modifier Hummers.
Effects of silver impregnation on bromide removal
The capacity of parent and Ag+ impregnated carbon adsorbents to remove Br− from water was evaluated in 100 mL batch bottle studies (25 mg/L adsorbents, 200 μg/L Br−). Figure 1 compares the percent removal of Br− and the Br− removal capacity of each adsorbent after 4 h of mixing. A control sample with no carbon recovered >99% of spiked Br−, which equates to <1% Br− removal in control experiments. The nonimpregnated adsorbents had minimal Br− removal (<3%). PAC-Ag, MH-Ag, and Tour-Ag removed 86%, 82%, and 91% of Br−, respectively.

Removal of spiked Br− (200 μg/L) in 1 mM NaHCO3 by 25 mg/L of adsorbents after mixing for 4 h. The white bars correspond to the left axis (% removal of Br−), and the gray bars correspond to the right axis (Br− removal capacity per mol of silver). The data represent average of experiment triplicates with error bars (total one standard deviation). Letters above bars indicate no statistical significance between data sets (one-way ANOVA, p < 0.05). ANOVA, analysis of variance; Br−, bromide.
Effects of background water characteristics on bromide removal
Figure 2 compares Br− removal by Ag+ impregnated carbon adsorbents for four different water matrices with controlled complexities. Tour-Ag removed >90% of Br− in all water matrices except natural CAP water, where it removed ∼75% of Br−, MH-Ag removed ∼70% of Br− when Cl− was introduced, ∼70% of Br− when Cl− and NOM were introduced, and it removed ∼50% of Br− in natural CAP water. PAC-Ag removed ∼60% of Br− when Cl− was introduced, ∼50% of Br− when Cl− and NOM were introduced, and only ∼30% of Br− in natural CAP water. We attributed the lack of Br− removal by PAC-Ag to its porous nature, where complex organics and competing ions can block pore channels or compete for sorption sites on the carbon, thereby rendering Ag+ not on the PAC-Ag surface unavailable for interactions with Br− in solution.

Removal of spiked Br− (200 μg/L) in four different water matrices by 25 mg/L of Ag+ impregnated PAC and GO after mixing for 4 h in polypropylene batch bottles. The water matrix chemistry is provided in the legend. The white bars correspond to the left axis (% removal of Br−), and the gray bars correspond to the right axis (Br− removal capacity per mol of silver). The data represent average of experiment triplicates with error bars (total one standard deviation). Letters above bars indicate no statistical significance between data sets (one-way ANOVA, p < 0.05). PAC, powdered activated carbon; GO, graphene oxide.
Jar testing experiments to simulate bromide removal at a water treatment facility
Figure 3 shows Br− removal for PAC-Ag, MH-Ag, and Tour-Ag in natural CAP water in jar tests simulating coagulation and flocculation in a water treatment facility. In each of the jar test experiments, the coagulant formed settleable flocs. Adding any form of silver achieved turbidity removal equal to the alum alone. Alum by itself did not remove Br−. In combination with alum, both MH-Ag and Tour-Ag removed ∼70% of Br−, while PAC-Ag with alum removed ∼40% of Br−, which was statistically worse than MH-Ag and Tour-Ag. MH-Ag with alum had the greatest Br− removal capacity per mol of Ag+ of the adsorbents.
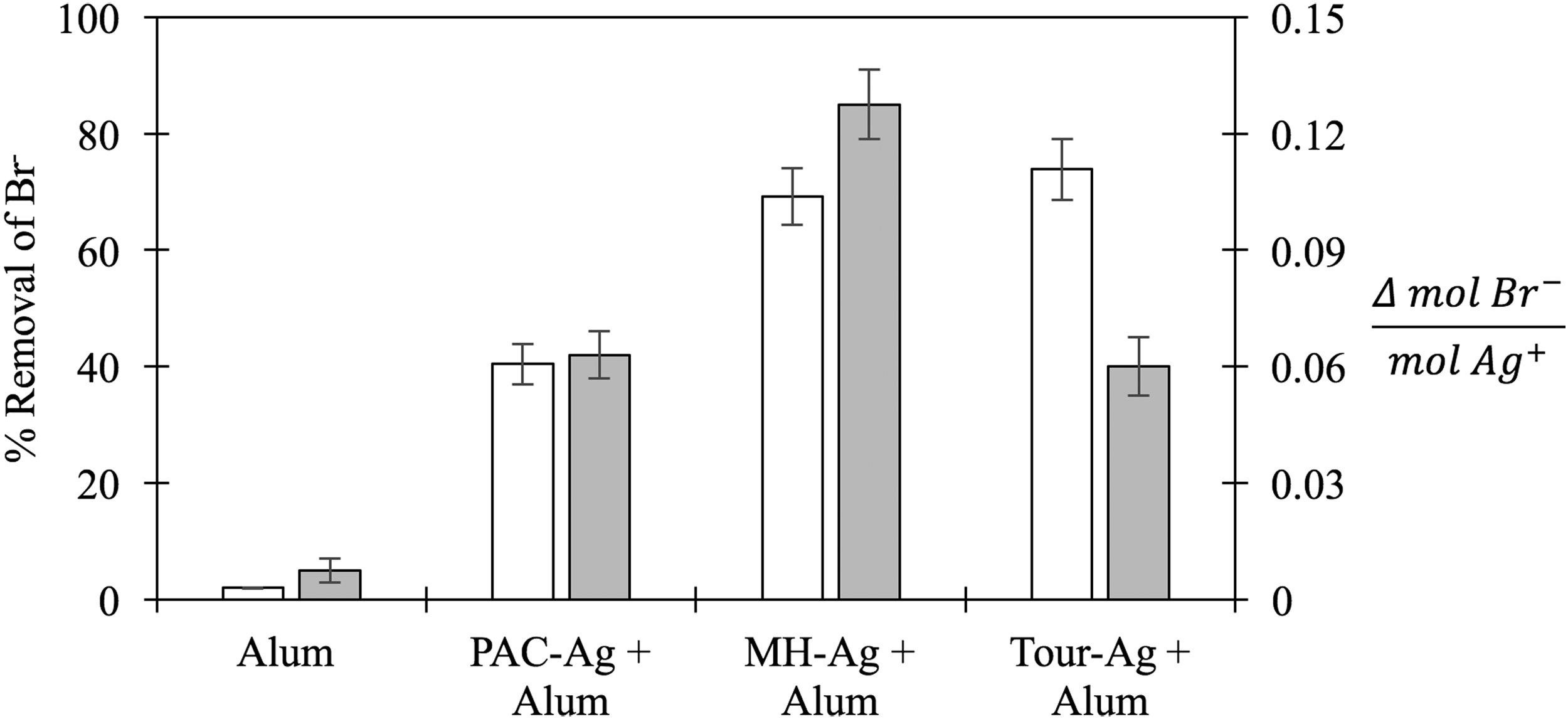
Removal of spiked Br− (200 μg/L) in natural Central Arizona Project water by 25 mg/L of Ag+ impregnated PAC and GO after jar tests with 28 mg/L alum. The white bars correspond to the left axis (% removal of Br−), and the gray bars correspond to the right axis (Br− removal capacity per mol of silver). The data represent average of experiment triplicates with error bars (total one standard deviation). Letters above bars indicate no statistical significance between data sets (one-way ANOVA, p < 0.05).
Discussion
Characterization of silver impregnated carbon sorbents
The silver impregnated adsorbent made with Tour-Ag had the highest Ag+ content by mass (12.6%) followed by PAC-Ag (5.8%) and MH-Ag (4.9%). This means Ag+ loading is not determined by the surface area of the material, as the lower surface area GOs had equivalent to higher silver content than PAC-Ag. Rather, the high Ag+ loading of the Tour indicates that Ag+ attaches to the oxygen groups found on the oxidized material. As GO oxygen content increased, more Ag+ was attached. Raman spectroscopy further confirmed the attachment of Ag+ to the carbon structure. The Raman spectra of the different materials after silver impregnation showed a consistent increase in the D band intensities (Supplementary Fig. S1). This increase is due to the adsorption of silver ions to the carbon lattice, thus disturbing the electron distribution of the material, as previously observed for silver nanoparticles or thiolated functionalized GO sheets (Das et al., 2011; Pham et al., 2013). This is not the case for PAC-Ag, which had less oxygen than the GO materials but slightly more Ag+ attached than MH-Ag. We believe that Ag+ attachment on PAC-Ag may be related to the SSA of PAC. To test this, the BET equation was used to calculate SSA before and after Ag+ impregnation. Ag+ surface attachment decreased the SSA of the adsorbents, with PAC-Ag having a significantly larger SSA than the GO, as expected. These findings align with previous findings, which were attributed to silver ions occupying sorption sites (Chen et al., 2016). Our work differs from prior work with silver impregnated PAC/GAC because we do not heat treat the material, which could form silver oxides (Chen et al., 2016). It is also different from the formation of zero valent silver, which requires H2O2 addition to produce Ag+ (He et al., 2012; Polo et al., 2016).
Effects of silver impregnation on bromide removal
Stoichiometrically, to remove 100% of the 200 μg/L Br−, there only needs to be 2.5E-06 moles of Ag+, which equates to 1.08% dry mass of Ag+ loaded on the tested carbon adsorbent dose of 25 mg/L; however, even with silver loadings of 4.9% (MH-Ag), 5.8% (PAC-Ag), and 12.6% (Tour-Ag), the maximum removal capacity of Ag+ for Br− is no higher than 0.13 mol Br− per mol Ag+. One explanation is that the high Ag+ loading on the carbon and the low Br− present in solution result in a high fraction of Ag+ unavailable to complexing with Br−. In Supplementary Figure S2, we compare Br− removal versus the initial Ag+ loaded onto the carbon adsorbents. Compared to the control (i.e., without Ag+), adding Ag+ increased Br− removal in all samples, but there was little difference in Br− removal with different Ag+ loadings, further validating that excess Ag+ to remove Br− is only beneficial to an extent. We attribute this to excess Ag+ in solution that may not come in contact with the small concentration of Br− available. These results indicated that (1) the impregnation of silver ions on the carbon adsorbents was required for Br− removal and (2) Br− removal capacity may not be influenced only by Ag+ loading onto carbon, but also the availability of silver for interactions with Br−.
Effects of background water characteristics on bromide removal
Br− removal capacity per mol of silver remained low for all three adsorbents across the four water matrices (<0.13 mol of Br− per mol Ag+). For both PAC-Ag and MH-Ag, their Br− removal capacity and percent Br− removal both decreased as chloride and NOM were added and as natural CAP surface water was used. We believe that this was a result of competing ions and organic matter complexing with the Ag+, rendering much of it unavailable to react with Br− in solution. For Tour-Ag, the amount of Br− removed per mol of Ag+ and the percent Br− removal both increased as chloride and NOM were added. In CAP surface water, the amount of Br− removed per mol of Ag+ was similar to the model water with Cl− and NOM; however, the percent Br− removal decreased. We attributed these results to the high silver loading on Tour-Ag (12.6%) relative to MH-Ag (4.8%) and PAC-Ag (5.9%). The high loading of Ag+ on Tour-Ag means that the Br− removal capacity per mol of Ag+ is relatively low in comparison to PAC-Ag and MH-Ag, both of which have half the amount of Ag+ present, when tested in model waters with limited competing elements. However, this high Ag+ loading on Tour-Ag means that more Ag+ is available to interact with Br− and competing ions present in more complex waters. This allows Tour-Ag to achieve greater Br− removal per mol of Ag+ and percent Br− removal than PAC-Ag and MH-Ag in CAP water. It is important to note that the increase in Ag+ present in Tour-Ag increases the potential for Ag+ and Br− interactions; however, competing ions will still have an effect on Br− removal.
Jar testing experiments to simulate bromide removal at a water treatment facility
Even though PAC-Ag had a similar Ag+ content to MH-Ag and it performed well when removing Br− from ultrapure water, intraparticle diffusion and kinetic challenges present in the complex natural water limit the ability of PAC-Ag to remove Br− in the natural CAP surface water. Instead, both MH-Ag and Tour-Ag showed greater promise for Br− removal in complex surface waters such as the natural CAP water tested. An additional test was done where we spiked AgNO3 salt at five times the stoichiometric ratio of Ag:Br into jar testers to determine if a carbon adsorbent was necessary for Br− removal. This test removed ∼50% of Br− and had a bromide removal capacity per mol of silver of <0.07 mol Br− per mol of Ag+ demonstrating that AgNO3 was less effective than silver impregnated carbon adsorbents at removing Br− from natural CAP water; however, this may be a result of the background characteristics of the natural CAP water, and results may vary with other surface waters.
Consideration of bromide removal mechanisms by Ag-impregnated GO
There are two potential mechanisms for Br− removal by Ag+ impregnated GO adsorbents: (1) Br− ions diffuse from the bulk water onto the sorbent surfaces where they complex with the Ag+ (Ag-Br); and (2) Ag+ releases from the adsorbents into water and interacts with Br− ions, forming sparingly soluble AgBr(s) crystals.
To determine if Br− complexes with Ag+ on the adsorbent surface, we captured PAC-Ag, MH-Ag, and Tour-Ag on 0.2-μm filters after Br− interactions and imaged them with SEM and EDAX. Figure 4 shows SEM images with and without EDAX elemental mapping of bromide, silver, chloride, and oxygen. Since EDAX provides information about the adsorbent surface, the silver that was not detected by EDAX should not be present on the adsorbent surface. For each of the adsorbents, most silver is in clusters on the adsorbent surface. Bromide was found on the filter surface and on the adsorbent surface and did not appear to cluster around silver or oxygen groups on the adsorbents. A control with Br− only (no adsorbents) showed that free Br− is retained by the filters, indicating that Br− found in the SEM/EDAX images not attached to carbon adsorbents is likely not free AgBr complexations. Chloride appears to be in clusters similar to silver clusters on the adsorbent surface, indicating that there may be complexation of AgCl on the carbon adsorbent surfaces. EDAX mapping has a detection limit of <0.1%, so more silver that is not detectable is likely present on the adsorbent surface. There was also some silver present on the filters, which may indicate free Ag+ in solution. ICP-MS data were collected for Ag+ in solution and verified that Ag+ leached off the adsorbents.
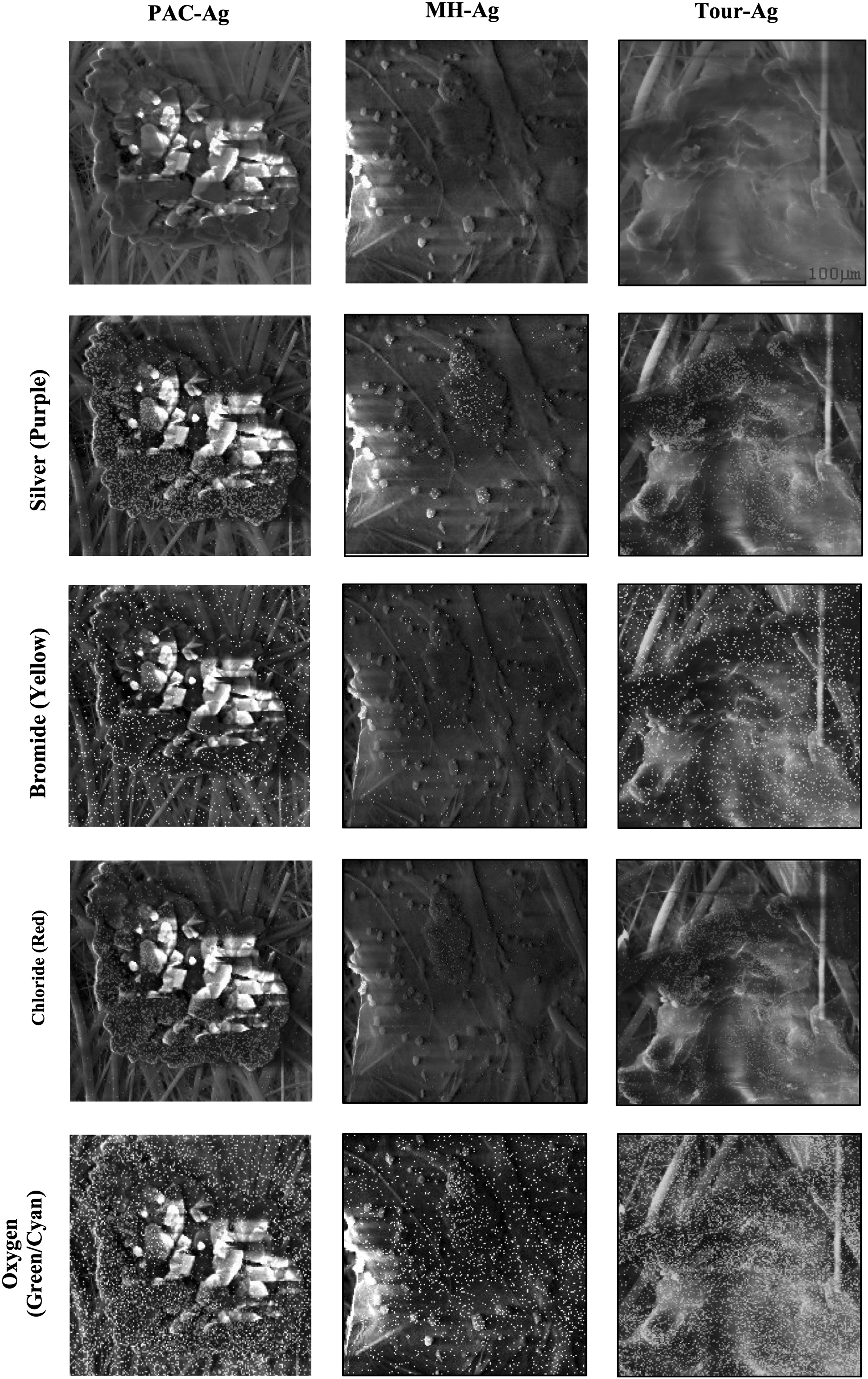
SEM images of PAC-Ag, MH-Ag, and Tour-Ag in 1 mM NaHCO3 with 200 μg/L bromide and 20 mg/L chloride. Each figure is on the same scale (top right figure). Each column corresponds to an individual adsorbent. The first row is of SEM images with no EDAX elemental scanning, and the other rows show silver, bromide, chloride, or oxygen from EDAX elemental scanning. The EDAX scans can be found in Supplementary Figure S3. EDAX, energy dispersive X-ray spectroscopy; MH, modifier Hummers; SEM, scanning electron microscopy.
To determine if Ag+ leaches into solution and forms AgBr complexations, we conducted a Ag+ leaching test for 1 mM NaHCO3 and Natural CAP water with and without Br− present. In Table 2, we explored the potential leaching of Ag+ into solution. When we compare the leaching of Ag+ in 1 mM NaHCO3 and CAP water in 1 mM NaHCO3 water, PAC-Ag and MH-Ag release 12.7 mg/g carbon and 9.9 mg/g carbon, respectively. In CAP water, PAC-Ag releases 3.6 mg/g carbon and MH-Ag releases 7.5 mg/g carbon. We observe that the amount of Ag+ leached into solution from PAC-Ag is more hindered by CAP water than MH-Ag. This demonstrates that the competing ions and NOM in CAP water prohibit the release of Ag+ into solution and is likely the reason why PAC-Ag performs worse than MH-Ag in complex waters.
Twenty-four hour acid digestion with nitric acid was conducted to approximate total silver content.
Natural CAP water refers to natural river water managed by the CAP.
PAC-Ag total silver content = 55.6 ± 11.7 mg Ag+/g carbon.
MH-Ag total silver content 49.2 ± 5.8 mg Ag+/g carbon.
Tour-Ag total silver content 126.2 ± 32.7 mg Ag+/g carbon.
Br−, bromide; CAP, Central Arizona Project.
When we compare the leaching of Ag+ in 1 mM NaHCO3 with and without Br− present, the amount of Ag+ present for all three adsorbents decreases significantly, indicating that the Ag+ that leaches into solution likely complexes with Br−, forming AgBr complexes and precipitating out of solution. This is also observed in Natural CAP water with and without Br− present. Lower amounts of Ag+ remain in CAP water than in 1 mM NaHCO3 water, indicating that Ag+ is also likely complexing with Cl− in solution, forming both AgBr and AgCl precipitates.
SEM/EDAX and silver leaching results illustrate that the likely potential mechanism for bromide removal by PAC-Ag, MH-Ag, and Tour-Ag is the leaching of Ag+ into solution and the complexation of AgBr precipitates. Additional future research is needed to determine the exact mechanisms at play for bromide removal by silver impregnated carbon adsorbents.
Environmental implications and research needs
Carbon adsorbents without silver impregnation were unable to remove Br− from surface water. In buffered ultrapure waters spiked with Br−, all Ag+ impregnated carbon adsorbents performed well; however, the introduction of competing ions (Cl−) and organics (NOM) and the use of natural CAP water significantly reduced the ability of PAC-Ag to remove Br− from surface waters, which we attributed to its porous nature. The sheet-like structure of MH-Ag and Tour-Ag provided the advantage of reducing the competition for adsorption sites compared to PAC-Ag. They performed as superior adsorbents for Ag+ based Br− removal. Both MH-Ag and Tour-Ag showed the ability to remove more Br− than PAC-Ag when competing ions and organics were present. All three silver impregnated adsorbents reduced Br− in surface water when used in conjunction with alum during coagulation and flocculation, making silver impregnated GO a viable technology to be introduced into the current treatment process framework of water treatment facilities. The mechanism for Br− removal by silver impregnated GO appears to be initiated by Ag+ leaching into solution, complexing with Br−, and forming and precipitating AgBr salts. In complex waters with chloride and/or NOM present, AgCl(s) or Ag-NOM complexes likely compete with AgBr reactions. The likely minor pathways for Br− removal are by Br− complexing with Ag+ on the adsorbent surface, which are shown in the SEM/EDAX images (Fig. 4).
Future research should address the need to improve the Br− removal capacity of Ag+ impregnated GO. Despite the improved performance of GO-Ag, Br− removal efficiency is still far from the theoretical molar ratio of Ag:Br of 1:1. To improve the economic viability of this new Br− removal process, future research should examine new materials and process designs that improve the Br− removal per amount of Ag+ added, as well as the recovery and regeneration of silver on these adsorbents. Changes in the material design, by changing the support structure or the silver impregnation process, are one possible development avenue.
Footnotes
Acknowledgment
This work was partially funded through the Nanotechnology-Enabled Water Treatment Nanosystems Engineering Research Center by the National Science Foundation (EEC-1449500).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.