
Abstract
Abstract
Effects of seasonal variation, including entirely different pollutant concentrations and temperature, on microbial community structures in a municipal wastewater treatment plant (WWTP) with anaerobic/anoxic/oxic (A2/O) system have been evaluated through high-throughput sequencing technique. Venn diagrams and bacterial diversity indices indicated significantly decreased biodiversity in winter. However, microbial community structure analysis showed high consistency among different A2/O chambers in summer or winter, while nearly all the dominant bacteria in summer changed in winter. Moreover, decreased ammonia-oxidizing bacteria (0.7% in summer and 0.3% in winter) coupled with low temperature fitted with the lower ammonia removal efficiency in winter. Functional genes predicted by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States indicated stability of their structures and contents in both summer and winter. These results suggested that seasonal variation would shift the microbial community structures, but not enough to change the functional genes. Results also provide a reference for the further study on microbial community structures of municipal WWTPs, especially with unstable performance.
Introduction
A
In addition, most of the existing researches revealed the differentiations of microbial community structures from the laboratory-scale bioreactors or among different WWTPs (Hu et al., 2012; Isanta et al., 2015; Chen et al., 2017). Microbial community structure changes with the operating conditions, which decides the removal efficiency of the pollutants in the biological treatment system. So analyzing and comparing the microbial community structures in WWTPs under different environmental conditions are indispensable to stabilize the effect, especially when this WWTP with unstable removal efficiency. A better understanding of the dynamics and function of the microbial communities under various conditions in these WWTPs will provide the first step for resolving this situation. However, the existing knowledge is insufficient to present the complex dynamics of the functional microorganisms and functional genes in these WWTPs. In this sense, the monitoring of the microbial communities combined with functional genes under various conditions is very important for the operation efficiency of biological treatment systems.
For a better understanding of the difference of microbial communities and functional genes along the process, we focused on a full-scale municipal WWTP with A2/O system, which cumulative treatment capacity ranked the first in China (accounting for 36.5%) (Xia et al., 2016). A total of six sludge samples from anaerobic, anoxic, and oxic chambers of A2/O in summer and winter were collected to analyze the microbial ecology under different environmental conditions. The microbial communities were explored by using 16S rRNA gene sequences and the functional genes were predicted by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt). Although functional genes in sludge samples are usually accurately studied by using quantitative real time PCR (qPCR) or metagenomics, PICRUSt is regarded as a novel functional genes prediction technique with high veracity in recent years (Wilkinson et al., 2018). The specific questions we address are the following: (1) what the microbial community structures of different chambers of A2/O system under different seasons are, and (2) what the predicted functional gene diversity and composition of A2/O system are. The results considerably extend our knowledge of the variations of microbial communities and functional genes in different processing units and seasons of full-scale WWTPs.
Materials and Methods
Location description and sampling
The full-scale municipal WWTP is located in Shenyang, Liaoning province of China, which treats domestic sewage at a scale of 20,000 m3/day. Other detailed information about the operational parameters and physicochemical conditions of the WWTP is summarized in Table 1. Daily measurement in duplicate was done to determine the pollutant concentrations in influent and effluent, which were collected by vertical point water sampler behind the sand collector and the clarifier, respectively. The temperature, pH, and dissolved oxygen (DO) concentrations of the oxic zone were also recorded twice when sampling, while the mixed liquor suspended solids (MLSS) concentration was done every 3 days. Throughout the experimentation period, the WWTP effectively removed 85% of chemical oxygen demand (COD) in the sewage; however, exhibited unstable ammonium (NH4+-N) removal efficiency (44.6–97%), especially low in winter (from December to the next February). In this study, the chemical analysis was done in July for summer and from November 2015 to December 2015 for winter. All activated sludge samples were collected in duplicate by vertical point water sampler at July 28 in summer and December 11 in winter from aerobic, anoxic, and oxic chambers, respectively. The two sludge samples from one point were thoroughly mixed and centrifuged at 4000 r/minute, and then they were stored in a −20°C refrigerator for further microbial community analysis.
COD, chemical oxygen demand; HRT, hydraulic retention time; SRT, sludge retention time; SS, suspend solid; TN, total nitrogen; TP, total phosphorus.
Analytical methods
Water samples were filtered through 0.45 μm pore size filters for water quality analysis. NH4+-N concentrations were measured in accordance with Standard Methods (APHA, 2005) on a spectrophotometer (SHIMADZU, Japan). MLSS and COD concentrations were also monitored by the method mentioned above. The DO and temperature of the wastewater were measured by portable hand-held DO meter (WTW, Germany).
DNA extraction and purification
For each sludge sample, DNA was first extracted from 2 mL sludge sample using a PowerSoil™ DNA Isolation Sample Kit (MoBio, USA), followed by the instructions given by the manufactures. Then the V3–V4 regions of the 16S rRNA genes were amplified with 341F (5′-CCT ACG GGN GGC WGC AG-3′) and 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′). The PCR amplification was done in GeneAmp 9700 Thermo Cycler (ABI, USA) and the process was as follows: 95°C preheating for 5 min, 94°C for 30 s (denaturation), 55°C for 30 s (annealing), 72°C for 1.5 min (extension) for 30 cycles, and 72°C for 7 min (final extension). The purified PCR amplicons were analyzed by an Illumina MiSeq platform at Sangon Biotech (Shanghai, China). The sequences were processed by quality control, in brief, adapters, barcodes, and primers in turn; sequences <200 bp and containing ambiguous “N” were removed from the raw sequences. The average length of contigs used in this study was 500 bp. Mothur 1.30.1 was used for computational analysis, and the bacterial reference database was SILVA. All the raw sequences involved in this study were deposited into NCBI's Sequence Read Achieve (SRA; accession no.: SRX3008166).
Functional genes predicted by PICRUSt
PICRUSt, a technique, was used to predict metagenomes from 16S data and a reference genome database (Langille et al., 2013). In this study, the National Center for Biotechnology Information's GenBank database was used. Functional predictions were categorized into Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Statistical analysis
Paired-sample t-test in the SPSS 19.0 software (Chicago, IL) was used to analyze the statistical significance in microbial community structures and function genes among different sludge samples. For summer or winter analysis, samples from different chambers were deemed as a replication. All values of tests are pointed out as means and standard deviations.
Results and Discussion
Bioreactor performance
Throughout the entire sampling period, the average COD and NH4+-N concentrations of influent and effluent are shown in Fig. 1. Influent concentrations of COD and NH4+-N in summer were 41.3–427.2 mg/L and 5.0–33.2 mg/L, respectively; the average concentrations were 161.58 mg/L and 21.62 mg/L. The dramatic change of influent concentration was due to the frequent rain in summer. While the influent concentrations of COD and NH4+-N changed more smoothly in winter, from 73.9–231.1 mg/L and 21.8–46.6 mg/L, respectively, the average values were 182.46 mg/L and 34.14 mg/L. The average COD and NH4+-N removal efficiencies were 81.6% and 86.5% in summer and 88.4% and 63.1% in winter, showing relatively stable treatment efficiency of COD and unstable of NH4+-N. Beyond that, temperature was another significant difference between summer and winter, for which the average values were 21°C and 11°C, respectively. Downing and Hopwood (1964) demonstrated that the process of nitrification can be limited by low temperature. Bioreactor pH, DO concentration, and MLSS were stably maintained at 7.2 ± 0.1 mg/L, 3.4 ± 0.2 mg/L, and 3300 ± 200 mg/L, respectively.
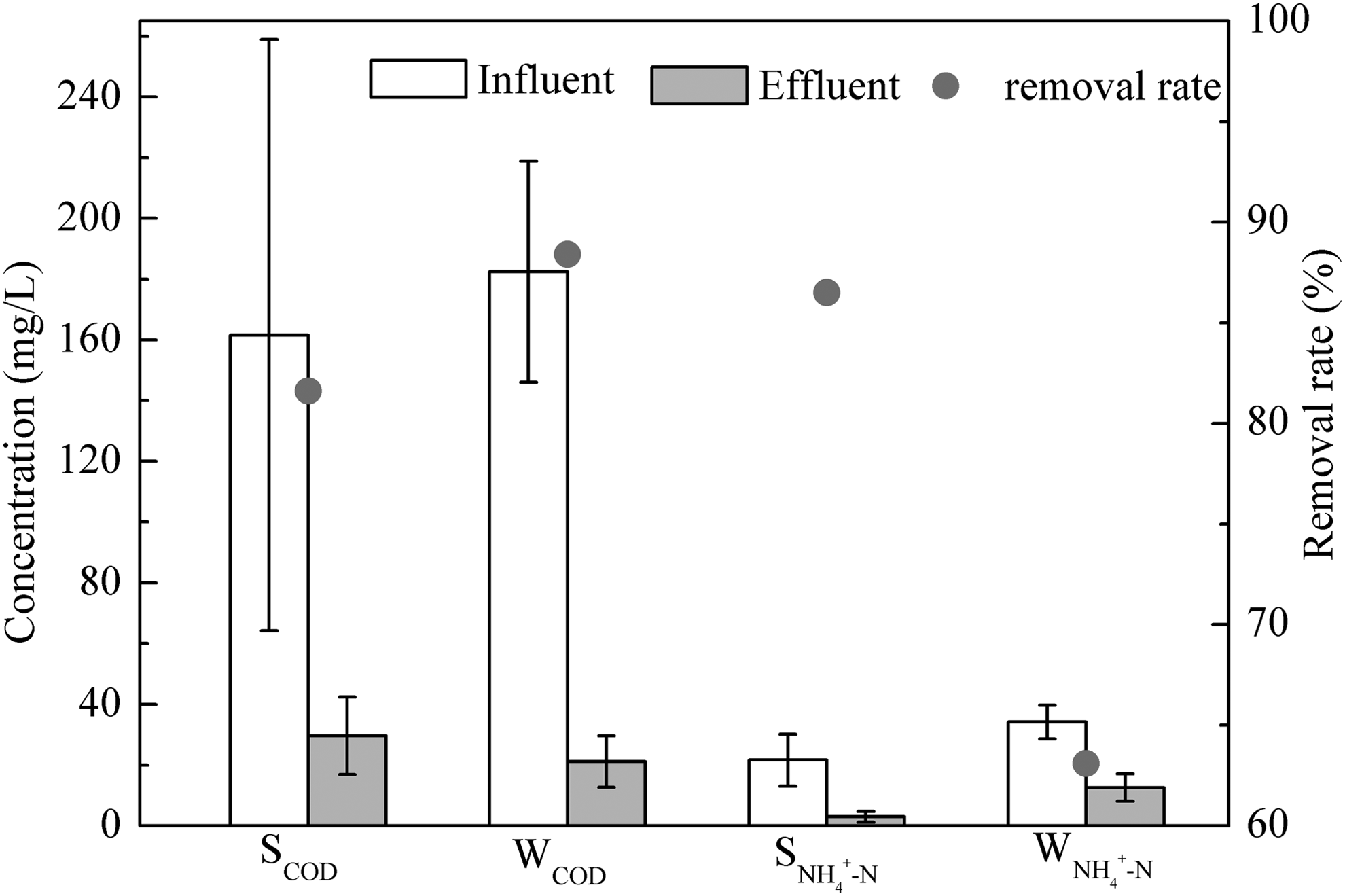
Average concentration of COD and NH4+-N in summer and winter. Error bars represent standard deviations. S and W stand for summer and winter, respectively. COD, chemical oxygen demand.
Bacterial diversity
A total of 6116 operational taxonomic units (OTUs) were detected, with 4568 and 4238 OTUs presented in summer and winter, respectively (Fig. 2), which indicated that the sludge samples in summer exhibited greater microbial richness than that in winter. The most part of OTUs were shared by anaerobic, anoxic, and oxic sludge samples, only 15.3% (699 OTUs), 14.4% (658 OTUs), and 14.0% (641 OTUs) of the total OTUs from summer were unique in anaerobic, anoxic, and oxic, respectively. Similarly, the number of unique OTUs from winter was only 14.4% (611 OTUs), 13.7% (582 OTUs), and 14.5% (616 OTUs) in anaerobic, anoxic, and oxic, respectively.

Venn diagram showing the overlap of operational taxonomic units in anaerobic versus anoxic, anaerobic versus oxic, and anoxic versus oxic from summer and winter. S and W stand for summer and winter, respectively; Y, Q, and H stand for anaerobic, anoxic, and oxic, respectively.
Diversity indices, including Shannon, Simpson, Ace, and Chao1, and coverage are listed in Table 2 at 97% similarity. The coverage of all samples ranged from 98.2% to 99.0%, demonstrating that the sequence libraries constructed in this study sufficiently covered the diversity of the microbial community (Chen et al., 2016). The greater microbial richness in summer than in winter was evidenced by Chao1 estimator of total OTU numbers and then validated by Ace indices. The Shannon–Wiener indices ranged from 6.11 to 6.22 in summer and 5.83 to 5.88 in winter, which acted as the Simpson indices, indicating that the microbial diversity in summer was higher in winter (Table 2). Different environmental parameters, such as the temperature and salinity, may result in the lower microbial diversity in winter (Du et al., 2011). The diversity indices of sludge samples from summer or winter were similar relative to the samples from different seasons.
Detected operational taxonomic unit number.
Shannon–Wiener index, higher number represents higher diversity.
Simpson indices, higher number represents lower diversity.
Ace index and eChao1 index, higher number represents higher species richness.
Microbial community structure
To reveal more information about the microbes, microbial community's composition of six samples at genus level is summarized in Fig. 3. To clarify the details of the bacteria communities of these sludge samples, partitioning ecological communities by their Bray–Curtis similarity was also shown in Fig. 3. The similarity decreased from higher to lower taxonomic levels, and the three samples (in anaerobic, anoxic, and oxic chambers) in summer or winter were divided into one group. A total of 450 genera were detected in these six sludge samples, and they, which relative abundance accounted for at least 90%, shared more than 69% genera. The overlap genera included unclassified bacteria, Dokdonella, Dechloromonas, and so on, which were also common genera shared by other WWTPs (Yang et al., 2011; Wang et al., 2016). However, their respective abundances were different among sludge samples, particularly in different seasons. Despite the shared genera, each season possessed some unique genera (relative abundance of 2.7% and 0.5% at most in summer and winter samples, respectively). These unique genera determine the diversity difference of microbial community structures.

Microbial community structures and similarity of sludge samples from different chambers in different seasons (S stands for summer and W for winter. Y, Q, and H represent sample from anaerobic, anoxic, and oxic chambers, respectively). Asterisk (*) and (**) indicate statistical significance between summer and winter at p < 0.005 and p < 0.001, respectively.
As shown in Fig. 3, the dominant microorganism, unclassified bacteria, accounted for at least 29.5% and 22.7% in summer and winter of total microorganism. The large percentage of unclassified species is the result of a large number of novel species inhabiting activated sludge communities (Zhang et al., 2016), which are ecologically significant in activated sludge process (Yang et al., 2011). Beyond unclassified bacteria (29.5, 31.6, 30.4%), Geothrix (3.0, 3.1, 2.9%), Intrasporangium (2.0, 2.6, 2.5%), Phaeodactylibacter (2.0, 1.9, 1.9%), Parcubacteria (2.1, 1.9, 2.1%), Aridibacter (1.9, 3.1, 2.6%), Terrimonas (2.6, 1.9, 1.9%), Jiella (1.8, 1.8, 1.8%), Gp7 (1.4, 1.6, 1.5%), and Tepidisphaera (1.6, 1.5, 1.2%) occupied great proportion of the samples in summer. The total abundance of these ten genera in summer took up 47.9, 51.0, and 48.8% in anaerobic, anoxic, and oxic chambers, respectively. The differences among all genera in these three chambers were less than 0.7% except the unclassified and Aridibacter, for which the largest differences were 2.1% and 1.2%. While, for the samples in winter, the ten most abundance genera were unclassified (22.7, 23.5, 23.4%), Dechloromonas (9.3, 8.2, 7.9%), Parcubactria (4.0, 4.1, 6.1%), Haliscomenobacter (3.6, 4.0, 4.2%), Phaeodactylibacter (3.2, 3.2, 3.3%), Rhodoferax (3.3, 3.2, 2.9%), Gemmobacter (2.2, 2.1, 2.1%), Dokdonella (2.3, 2.1, 1.7%), Saccharibacteria (1.8, 1.6, 1.4%), and Sideroxydans (1.3, 1.1, 1.4%). The total relative abundances of these genera were 53.7, 53.1, and 54.4% in anaerobic, anoxic, and oxic chambers, respectively. The largest difference of 2.1% occurred in Parcubacteria, which detected in oxic sample was surprising since this genus has been described as exclusively anoxic (Harris et al., 2004). The Parcubacteria are ectosymbionts or parasites of other organisms, and none of these species has been isolated in the laboratory (Nelson and Stegen, 2015). However, the alternating anaerobic and aerobic units of WWTP provide different microenvironments that could harbor these disparate microorganisms. Besides these genera previously mentioned, other genera have smaller amount and fewer differences among the different chambers.
The species of microbes and their relative abundance clearly showed high similarity among sludge samples from different chambers of A2/O system, although the largest differences occurred in unclassified and Parcubacteria (only 2.1%) in summer and winter, respectively. Sludge return and internal recycle (70% and 200%, respectively) of A2/O system cause the consistency, while sequencing depth and coverage in different samples may result in these differences (Sims et al., 2014).
According to the above results, the sludge samples of anaerobic, anoxic, and oxic in summer or winter were considered as replication in analyzing the difference between summer and winter. As anticipated, considerable similarities and differences (e.g., unclassified, the dominant bacteria all the time, with different relative abundance) were shown in the microbial community structures between summer and winter, because the different environmental conditions, including temperature and concentrations of pollutions, would be different for the microbial communities (Akyol et al., 2016). In addition, the change of microbes mainly occurred in the core bacteria, for example, unclassified bacteria (30.5 ± 1.1%), Geothrix (3.0 ± 0.1%) and Aridibacter (2.5 ± 0.6%) were the average top three dominant genera in summer. Meanwhile, these three genera also existed in winter; however, their abundances were much lower. In addition to the three dominant genera mentioned above, Intrasporangium, Gp7, Exiguobacterium, Pirellula, Jiella, and so on were abundant in summer (1.4–2.4%), while their abundances also decreased in winter (0–0.6%). In previous studies, some of them were quite sensitive to the environment, for instance, Geothrix and Gp7 (Yang et al., 2014). The abundance with the steepest drop in winter was unclassified bacteria, as indicated by the bacteria diversity (Table 2). Another remarkable genus, Exiguobacterium, disappeared entirely in winter (1.4 ± 0.3% in summer). Exiguobacterium is a kind of antibiotic-resistant bacteria (Tahrani et al., 2015), the abundance depends on the concentration of the antibiotics. Moreover, the average top three dominant genera in winter were unclassified bacteria (23.2 ± 0.4%), Dechloromonas (8.5 ± 0.7%), and Haliscomenobacter (3.9 ± 0.3%). Among all of the genera, the abundance of Dechloromonas had the largest variation, average ranging from 0.3% in summer to 8.5% in winter. Dechloromonas was known to oxidize aromatic compounds (Drury et al., 2013), their higher abundance may reflect higher influent concentration of polycyclic aromatic hydrocarbons in winter (Qiao et al. 2014). It is also worth noting the increasing of Haliscomenobacter in winter and Haliscomenobacter playing an important structural role in sludge floc formation that can also cause serious sludge bulking when they are overproliferating (Kotay et al., 2011).
Based on the analysis of microbial community structures from the anaerobic, anoxic, and oxic in summer and winter, it could be concluded that the change of season (difference of influent and temperature) primarily affected the microbial community relative abundance, but had limited influence on the microbial species.
Key nitrogen removal microorganisms
Biological nitrogen removal is one of the most important functions in this WWTP, including nitrification and denitrification (Yu and Zhang, 2012). As the first and the limiting step of nitrogen removal, nitrification is a biological oxidation of NH3 to NO3−-N (Ma et al., 2013), the involved microorganisms include AOB and NOB. For further analyzing the functional microbes of nitrification in the WWTP, the AOB and NOB in summer and winter were specifically analyzed. Among the WWTP samples, the species composition of AOB and NOB in different seasons was stable, despite the abundance changes with seasonal variation (Fig. 4). These changes were determined by the available substrate and various abiotic factors such temperature, pH, salinity, and presence of micronutrients (Nielsen et al., 2010). Most of the AOB detected in WWTP at both seasons belonged to Nitrosomonas, followed by Nitrosospira. There were sequences representative of Nitrospira, Nitrobacter, and Nitrolance, members of the NOB. In concordance with previous studies, Nitrosomonas and Nitrospira were the dominant AOB and NOB, respectively, in the WWTP (Whang et al., 2009), which played an important role in the nitrification process. A very interesting phenomenon was that the ammonia removal rate decreased in winter (Fig. 1), while the abundance of NOB increased, from 0.6 ± 0.2% in summer to 0.9 ± 0.1% in winter. Low temperature in winter may improve the amount of Nitrospira (Siripong and Rittmann, 2007). In previous studies, oxidation of ammonia to nitrite was confirmed as a rate-limiting step of the full nitrification process (Ma et al., 2013), so the decreased abundance of AOB in winter (0.7 ± 0.23% in summer; 0.3 ± 0.03% in winter) and changes of environmental conditions such as a decrease in temperature directly resulted in the low ammonia removal efficiency (Khunjar et al., 2014).
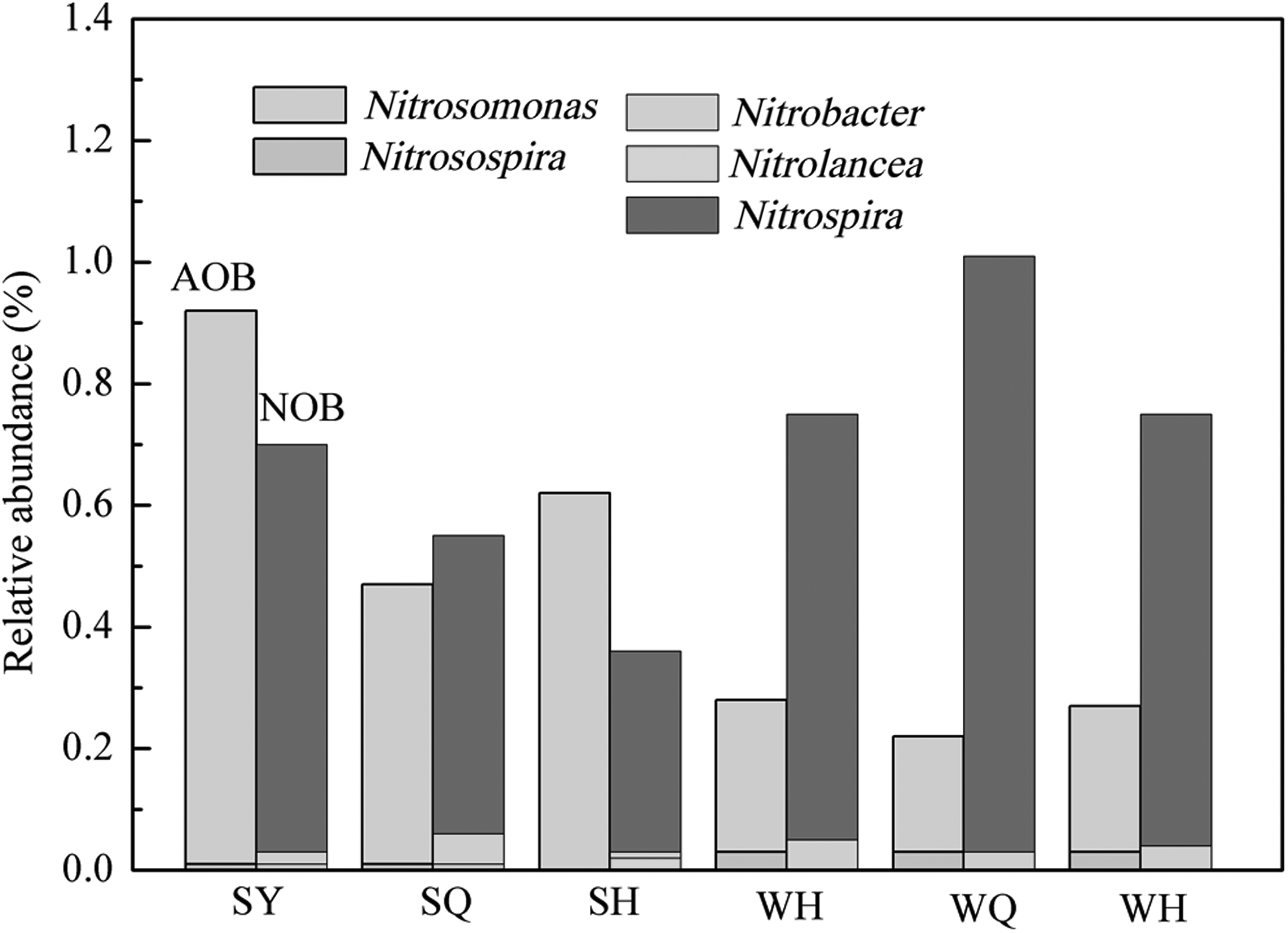
Relative abundance of AOB and NOB in different chambers from summer and winter. AOB, ammonia-oxidizing bacteria; NOB, nitrite-oxidizing bacteria.
Global functional gene classification
To clarify the details of functional gene of the activated sludge in this full-scale WWTP, functional gene analysis was also conducted by PICRUSt in this study. Compared to biology analysis, functional gene prediction by PICRUSt has the features of both accuracy and low cost (Langille et al., 2013).
A total of 6909 genes were detected, with 5799 genes in summer and 5507 genes in winter. These genes were involved in 41 gene categories level 2 KEGG Orthology groups (KOs) in GenBank dataset. Figure 5 shows the relative abundance of gene categories within the sludge metagenomes. It is easy to see that the same microbial community proved the same functional genes. Therefore, the functional genes of sludge samples from anaerobic, anoxic, and oxic in summer or winter were regarded as replication because of their consistency in microbial community structures (Fig. 3). The functional gene categories of sludge samples in summer and winter are extremely similar with each other, regardless of the tremendous differences in microbial community. Results of KEGG annotation analysis showed that average 50.9 ± 0.1% and 50.2 ± 0.1% of the genes detected in the WWTP were related to metabolism, 16.8 ± 0.1% and 16.7 ± 0.1% to genetic information processing, and 14.1 ± 0.1% and 14.4 ± 0.04% to unclassified in summer and winter, respectively. Only 1.8 ± 0.003% and 1.8 ± 0.007% annotated sequences were involved in the pathways of human diseases and organismal systems in summer and winter. In the category of metabolism, amino acid (10.6 ± 0.03% and 10.6 ± 0.02%), carbohydrate metabolism (10.1 ± 0.02% and 9.7 ± 0.01%), and energy metabolism (6.2 ± 0.03% and 6.1 ± 0.07%) have absolute advantage in both seasons. These genes ensure successful growth and metabolism pathway of the microbial communities in sludge. Similar to the findings about soil (Wu et al., 2016), amino acid metabolism and carbohydrate metabolism were the most two abundant categories in all sludge samples, because of their housekeeping functions of all living organisms. Overall, almost all the pathways in this study were ubiquitous for activated sludge (Gao et al., 2016).

Relative abundance (%) of predicted functional genes found in different chambers from summer and winter. S and W for summer and winter, respectively; Y, Q, and H for anaerobic, anoxic, and aerobic, respectively. Asterisk (*) and (**) indicate statistical significance between summer and winter at p < 0.005 and p < 0.001, respectively.
Influent supply more carbon substrate in winter as shown in Fig. 1, however, the genes related to carbohydrate and energy metabolism were found to be slightly lower in winter (10.1 ± 0.02% and 6.2 ± 0.03% in summer; 9.7 ± 0.01% and 6.1 ± 0.08% in winter), which do not conform to the higher resource availability. This contradiction may be explained by different growth rate of microbe species and selectivity absorption of carbon sources by microbes. The other obvious difference between summer and winter was the abundance of xenobiotics biodegradation and metabolism (2.9 ± 0.01% in summer and 3.2 ± 0.07% in winter), which tended to be higher in winter. Xenobiotics biodegradation and metabolism were pathways to eliminate or inactivate the toxic substrates (Lee et al., 2015). Higher concentration of polycyclic aromatic hydrocarbons in winter (Qiao et al. 2014) may result in this difference.
Due to the differences in environmental conditions between summer and winter (e.g., temperature and concentrations of pollutions), significant differences of functional genes were expected to occur in different seasons. However, the most striking difference of gene category between them was carbohydrate metabolism, with a difference of 0.4%. Therefore, considering the gene categories and their abundances, the functional genes were much more stable than the microbial community structures between summer and winter. The possible cause of this phenomenon is that the fundamental metabolism highly shared by microbes, and the external environmental conditions are not enough to make significant variation in functional genes.
Environmental conditions, for example, temperature, salinity, and concentrations of pollutions, will keep the microbial community structures distinct; however, the conditions that can affect functional genes need further study. Meanwhile, the structures of microbial community and functional genes under different operating conditions and sewage treatment systems need further research, especially on the accurate functional genes by qPCR or metagenomics.
Conclusion
COD removal efficiency in WWTP was stable both in summer and in winter, while NH4+-N removal efficiency was affected by low temperature in winter (only 63.1%). Microbial community structure analysis showed that there is a high similarity among the sludge samples from the three chambers of A2/O system, despite the seasonal changes. However, the bacterial diversity significantly decreased in winter, and great differences in the microbial community structures were observed between summer and winter. Nitrosomonas and Nitrospira always were the dominant AOB and NOB in the WWTP, and the decrease of AOB abundance (0.3%) and temperature (11°C) in winter contributed to the lower ammonia removal efficiency. The functional genes in every category were fairly stable, regardless of the different environmental conditions.
Footnotes
Acknowledgments
The authors acknowledge the Major Science and Technology Program for Water Pollution Control and Treatment of China (Grant No. 2012ZX07202-010) for their financial support and Sangon Biotech (Shanghai, China) for their technology support.
Author Disclosure Statement
No competing financial interests exist.