
Abstract
The frequent occurrence of per- and polyfluoroalkyl substances (PFAS) in the water environment is a recent global concern. Because of their extraordinarily strong chemical stability caused by the multiple C–F bonds, decomposition of PFAS by using relatively practical technologies is the most challenging but significant task. In this context, this study focused on evaluating the potential of zerovalent iron (ZVI) particles combined with common oxidants such as hydrogen peroxide, persulfate, and peroxymonosulfate for the removal of perfluorooctanesulfonic acid (PFOS). Oxidant type and dose, temperature, and pH were controlled to generate various oxidizing and reducing reactive species separately or in combination. In most of the cases, a substantial amount of PFOS was removed. However, building up of identifiable expected intermediates and fluoride ions in water was not significant, implying that the observed PFOS removal could be ascribed to presumably its adsorption to and/or complexation with iron (Fe) species such as Fe oxides and dissolved Fe ions originating from ZVI. To confirm the persistence of PFOS, perfluorooctanoic acid (PFOA) with the very similar structure to PFOS was briefly examined. PFOA was well decomposed, producing many expected intermediates such as shorter chain PFAS. The results indicated PFOS, unlike PFOA, seems much more stable even in the presence of the strong reactive species produced by the integrated system.
Introduction
Per- and polyfluoroalkyl substances (PFAS) used in many industrial and military sectors and consumer products in our daily life, such as nonstick cookware, food packaging, stain and water-resistant fabrics, and aqueous film forming foams, have greatly contaminated the environment, particularly drinking water resources (Kissa, 1994; Houtz et al., 2013). Many of PFAS released into the environment undergo biological and environmental transformations eventually to the most oxidized state, that is, perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) (Dasu et al., 2013; Eriksson et al., 2017). Because of their widespread uses and bioaccumulative properties, PFOS and PFOA have been the most studied PFAS. The United States Environmental Protection Agency has set an advisory level in drinking water at 70 ng/L for them, and has designated PFAS as national priorities (USEPA, 2016, 2018).
The carbon chain in PFOS and PFOA is surrounded by fluorine and attached to a functional group at the end, creating the hydrophobic and oleophobic properties beneficial to many commercial products. The C–F bonds that PFAS contain (i.e., 17 in PFOS and 15 in PFOA) are known to be one of the strongest single covalent bonds in organic chemistry (note Fig. 8 later for the chemical structure of PFOS and PFOA). The C–F bonds are highly electronegative in nature, and cannot be oxidized easily (Vecitis et al., 2009; Merino et al., 2016). Most of PFAS are known not to be decomposed by conventional water treatment processes (Appleman et al., 2014). Advanced oxidation technologies (AOTs), which generate and exploit strong transient species such as hydroxyl radicals (•OH), sulfate radicals (
However, previous studies on AOTs showing successful decomposition of PFAS commonly used an external energy source, such as heat, ultraviolet, ultrasound, microwave, and electron beam, to diminish the structural integrity of PFAS, rather than focusing solely on the generation of radical species for promoting chain reactions (Hori et al., 2004, 2005, 2007; Moriwaki et al., 2005; Dillert et al., 2007; Lee et al., 2009, 2010; Panchangam et al., 2009; Rayne and Forest, 2009; Park et al., 2011; Lyu et al., 2015). Among many AOTs, reactive species can be most practically generated through the Fenton (or Fenton-like) reactions, where transition metals such as iron (Fe) are used as a catalyst to activate oxidants including hydrogen peroxide (HP or H2O2), persulfate (PS or
Meanwhile, reduction of C–F bonds in PFAS might be thermodynamically possible. Previously, zerovalent iron (ZVI) as a reducing agent has been popular to reductively decompose and thus dehalogenate many halogenated chemicals such as trichloroethylene and polychlorinated biphenyls (Choi et al., 2008; Fu et al., 2014; Lefevre et al., 2016; Zhang et al., 2017). However, no reports have shown successful defluorination of PFAS using ZVI alone under ambient conditions. Modification of ZVI or introduction of other similar mechanisms has been studied to improve the reaction kinetics (Arvaniti et al., 2015; Lawal and Choi, 2018). However, direct evidences for defluorination were not reported. When PFOS was subjected to extreme conditions (e.g., subcritical water at 350°C), maximum reduction of PFOS was observed in the presence of ZVI (Hori et al., 2006). In general, chemical reduction or defluorination, even if occurring under extreme conditions, results in defluorinated chemicals as final products that are more vulnerable to further chemical reactions through oxidation mechanisms.
Consequently, there have been efforts to generate various oxidizing and reducing species in combination to accelerate the decomposition of PFAS (Trojanowicz et al., 2018). In our recent study, successful decomposition of PFOA was achieved by combining oxidants and Fe-modified diatomite, where various radicals such as
In this study, we conjugated ZVI with common oxidants such as HP, PS, and PMS. ZVI as a strong reducing component released Fe2+ ions that activated added oxidants while generating various oxidizing and reducing reactive species including free electrons, di-hydrogens,
Experimental
Chemicals and reagents
PFOS (heptadecafluorooctanesulfonic acid potassium salt, C8F17SO3K, CAS 2795-39-3), PFOA (pentadecafluorooctanoic acid, C8F15O2H, CAS 335-67-1), ferrous sulfate heptahydrate (FeSO4·7H2O), ferric sulfate pentahydrate (Fe2O12S3·5H2O), ferrous oxide (FeO), ferric oxide (Fe2O3), sodium borohydride (NaBH4), sodium persulfate (Na2S2O8), potassium peroxymonosulfate (KHSO5·½KHSO4·½K2SO4), sodium hydroxide (NaOH), and hydrochloric acid (HCl) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN or C2H3N), ethanol (C2H6O), methanol (CH4O), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), formic acid (CH2O2), acetic acid (C2H4O2), and H2O2 (30% in weight) were acquired from Thermo Fisher Scientific (Waltham, MA). All solutions and sample preparations were carried out in ultrapure water (18 MΩ·cm) produced by a Millipore Milli-Q filtration system (Billerica, MA). Commercially available ZVI, named reactive nanoscale iron particle (RNIP), was purchased from Toda Kogyo (Yamaguchi, Japan). All chemicals were used as received. Large molecule separation (LMS; 200 mg, 3 mL) and strong cation exchange (SCX; 200 mg, 3 mL) cartridges for solid phase extraction (SPE) were purchased from Agilent Technologies (Santa Clara, CA).
Synthesis and characterization of ZVI
ZVI (boron-coated Fe) was synthesized through borohydride reduction of ferrous ions as described elsewhere (Liu et al., 2006). In brief, 3 g ferrous sulfate was dissolved in 4:1 (v/v) ethanol:water solution (90 mL) and mixed in a rotary shaker at 120 rpm to which 150 mL of 0.1 M NaBH4 was added dropwise. ZVI was recovered by filtering the slurry with 0.45 μm glass fiber filter and washed with ethanol to remove any residual chemicals followed by CH4O to prevent ZVI from oxidation. ZVI was submerged in ethanol or CH4O during its synthesis and stored in an airtight vial; thus, oxidation of ZVI before its use was minimized. Then, ZVI was briefly characterized to confirm its properties. X-ray diffraction (XRD) using a Kristalloflex D500 diffractometer (Siemens, Munich, Germany) was used to investigate the crystallographic properties. The morphology was investigated with a high-resolution transmission electron microscope (HR-TEM; JEOL JEM-2010F, Akishima, Tokyo). The size was determined using a SZ-100 particle size analyzer (Horiba Scientific, Irvine, CA). The surface area was determined using a Tristar 3000 (Micromeritics, Norcross, GA) porosimetry analyzer.
Batch experiment
Reactions were carried in airtight 20 mL polypropylene reaction vials containing a final volume of 20 mL with 10 mg/L PFOS (0.02 mM). Concentrations of oxidants were 0.3 M for PS and PMS, and 1.5 M for HP. Because salts such as PS and PMS greatly increase the ionic strength of the solution and pose analytical challenges for PFOS, PS and PMS were used in much lower concentrations than HP. These concentrations were also adopted from previous studies using oxidants to decompose similar target compounds (Mitchell et al., 2014; da Silva-Rackov et al., 2016; Wang and Wang, 2018). ZVI concentration was 0.5 g/L. Reaction temperature was fixed at 20°C unless otherwise mentioned. Initial pH was controlled using 2 M HCl or 2 M NaOH (in most of the tests, NaOH was needed to control pH because addition of the reactants decreased pH significantly). Buffer species were not used because they were expected to react with the radicals and to interfere with the analysis of PFOS. To investigate the effects of operational parameters, oxidant doses were varied at 0.03 and 0.3 M for PS and PMS and 0.15 and 1.5 M for HP; temperature was controlled at 20°C, 40°C, and 60°C; and initial pH was controlled at 3 (acidic), 7 (neutral), and 9 (basic). One experiment on a comparison basis to PFOS was conducted using PFOA under the same conditions. The reaction time for the batch setup was prolonged up to 48 h to observe possible reaction intermediates from the decomposition of PFOS and PFOA, if any.
PFOS removal potentially by adsorption to solid Fe2O3 and FeO particles under different initial pHs of 3, 7, and 9 was also attempted with 2 g/L Fe2O3 and 0.8 g/L FeO that resulted in 0.5 g/L as Fe. Instead of the Fe oxides, dissolved Fe3+ and Fe2+ ions were added to check for the possible complexation of PFOS with the Fe species. A proper amount of FeSO4·7H2O and Fe2O12S3·5H2O salt was added to achieve 0.25 − 2 g/L Fe3+ and Fe2+ under pH 3.
Sample treatment and chemical analysis
Samples of 0.2 mL were diluted in 1.8 mL of ultrapure water. LMS cartridges were preconditioned using 12 mL of CH4O and washed with 12 mL of ultrapure water. Later, samples were extracted with CH4O in a ratio of 1:1. The samples were analyzed in a Shimadzu (Nakagyo-ku, Kyoto, Japan) Nexera liquid chromatography (LC) equipped with a Shimadzu 8040 triple quadrupole mass spectrometer (MS). The flow rate was set at 0.8 mL/min with a binary gradient method. Mobile phase was 0.1% CH2O2 in water and in ACN. Separation was achieved by an Agilent Zorbax Eclipse Plus C18 column (1.8 μm particle size), and target analytes were eluted through a gradient method in which ACN was increased 5–100% over 6 min, kept at 100% for 3 min, and then brought down to 5% in 1 min. Column temperature was set at 40°C, and sample injection was set at 10 μL. For quantification of the target analyte such as PFOS and PFOA and expected byproducts such as short-chain PFAS, multiple reaction monitoring (MRM) scans were conducted in a negative electrospray ionization mode. Confirmation of expected reaction intermediates was undertaken using targeted analysis. Monitored ion transitions of PFOS, were conducted as described by Bruton and Sedlak (2017).
To find any changes in the molecular structure of PFOS, particularly by defluorination, nuclear magnetic resonance spectrometry (NMR) analysis was carried out in a 300 Hz Oxford instrument (Abingdon, Oxfordshire, United Kingdom). PFOS and intermediates were extracted using CH4O in 1% C2H4O2. Then, sample was evaporated using a rotovamp and later dissolved in 0.5 mL deuterated CH4O. For the identification of fluoride ions detached from PFOS, ion chromatography was used using a Dionex (Thermofisher, Waltham, MA) DX-500 system (theoretical concentration of fluoride ions in water, when fully released, was 6.1 mg/L). SPE was conducted with SCX cartridges to extract fluoride ions. These cartridges were conditioned with 3 mL CH4O and equilibrated with 3 mL ultrapure water before loading 0.5 mL of the sample, which was then collected for analysis. The column used for the separation was a Dionex Ionpac AS-14A (4 × 250 mm) coupled with a Dionex Ionpac guard column AG-14A (4 × 50 mm). The eluent was 4.5 mM Na2CO3 and 0.8 mM NaHCO3. An isocratic method with a flow rate of 1 mL/min was applied. A 4 mm Dionex AERS suppressor was used to reduce background conductivity of the mobile phase.
Results and Discussion
Reactivity of ZVI (RNIP and ZVI)
Before the batch experiment, we briefly characterized the homemade ZVI (herein, ZVI) synthesized through the well-established method to confirm that Fe particles in the samples are in zerovalent state and at nanoscale (trivial graphical data are not provided in this text). XRD results indicated the presence of ZVI at 2θ of 45°. HR-TEM showed ZVI nanoparticles in size of ∼19 nm and RNIP in size of ∼45 nm. Surface area of ZVI was 37.5 m2/g, whereas that of RNIP was 30 m2/g. The properties were very close to those reported elsewhere (Liu et al., 2006; Choi et al., 2008).
Because hydrodehalogenation capability of ZVI has been well studied with persistent chemicals such as polychlorinated biphenyls (Choi et al., 2009a), the reactivity of both RNIP and ZVI was examined for defluorination of PFOS under standard conditions, as given in Fig. 1a showing aqueous concentration change of PFOS. RNIP was not able to significantly remove PFOS through reductive defluorination or even physical adsorption. Although the unique homemade ZVI, called boron-coated Fe, has been known to generate highly reactive di-hydrogen through reduction of water, which is involved in strong hydrodehalogenation reaction, no PFOS was removed. Accordingly, no fluoride ions were detected in the reaction samples, suggesting that ZVI alone lacks the capability to reductively defluorinate PFOS under the tested conditions. Although dehalogenation, particularly dechlorination by ZVI or many other zerovalent metals, has been thoroughly used in environmental applications, defluorination of PFOS might be more challenging because of the absence of low-laying vacant d orbitals to accept an electron in PFOS. Recent studies also pointed out that defluorination of PFAS by ZVI-based materials is thermodynamically possible but kinetically negligible (Blotevogel et al., 2018; Lawal and Choi, 2018; Park et al., 2018).

Reactivity of oxidants
To examine if oxidants alone can remove PFOS through the conventional oxidation pathway, the reactivity of each oxidant with PFOS was examined under standard conditions, as given in Fig. 1b. Although HP, PS, and PMS have been widely used as strong oxidants to decompose a variety of chemicals (Bennedsen, 2014), none of them were able to decompose PFOS. The result was expected from the strength of the C–F bonds in PFOS and the absence of C–H bonds vulnerable to chemical reactions with the oxidants.
It is known that oxidants can be activated more vigorously at high temperatures. Specifically, heat-activated PS has been successful in degrading many organic chemicals, whereas other oxidants such as PMS and HP have been reported to be less or not affected by reaction temperature (Yang et al., 2010). The effect of temperature at up to 60°C on the reactivity of oxidants with PFOS was tested, as given in Fig. 2. Reaction time was expanded to 3 h and initial pH at 3.0 evolved to ∼1.5 in the end. Of interest, none of the conditions showed meaningful removal of PFOS. Decomposition by heat-activated PS (80°C, pH <3) has been reported exclusively for PFOA, whereas decomposition of PFOS has been reported at much lower or negligible rates (Bruton and Sedlak, 2018). Similarly, Park et al. (2016) also reported that PS alone under high temperatures oxidized PFOA but not PFOS. Oxidant activation temperature does not seem to impact on the decomposition of PFOS.
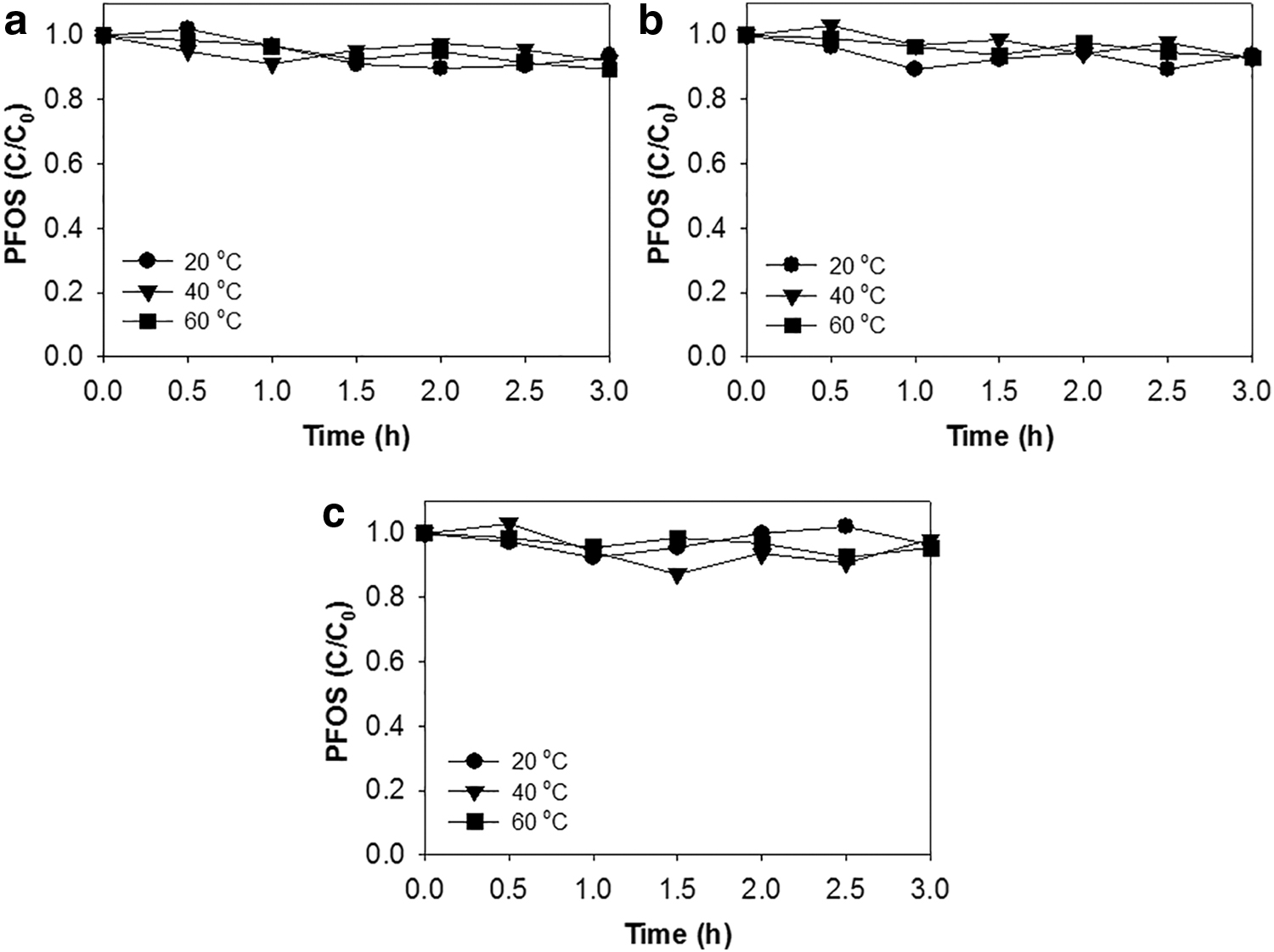
PFOS removal by oxidants alone
ZVI conjugated with oxidants
Because ZVI alone for defluorination or adsorption of PFOS and oxidants alone for oxidation of PFOS did not work, ZVI was conjugated with oxidants as given in Figs. 3–5 to produce various oxidizing and reducing radicals including •OH,

PFOS removal by ZVI conjugated with oxidant
PFOS removal by ZVI conjugated with oxidant

PFOS removal by ZVI conjugated with oxidant
The ZVI-based integrated system was able to remove PFOS. As given in Fig. 3 (note the result only at 20°C at this moment), PFOS removal was marginally improved with the integrated systems compared with oxidants or ZVI alone showing negligible removal of PFOS. The integrated systems of PS, PMS, and HP in the presence of ZVI presented 7%, 8%, and 12% PFOS removal, respectively.
Effects of temperature, oxidant dose, and reaction pH
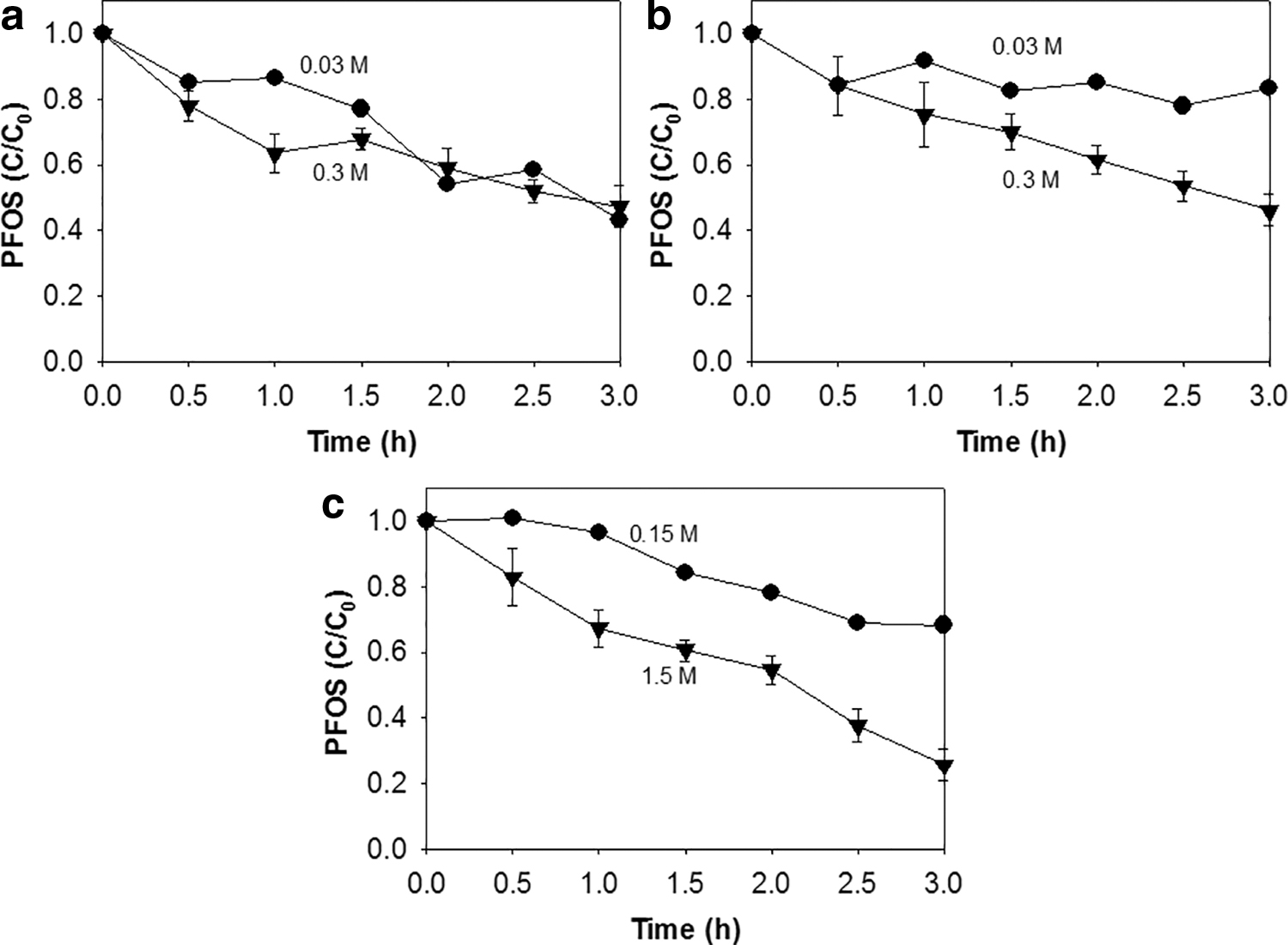
Reaction conditions were adjusted to accelerate the PFOS removal kinetics and to elucidate the effects of operational parameters. Increased reaction kinetics was also believed to help to identify reaction intermediates and fluoride ions produced as a result of PFOS decomposition and thus to interpret any possible removal mechanisms. ZVI conjugated with PS was examined at different temperatures, as given in Fig. 3a. Of interest, PFOS removal was much improved from 7% at 20°C, 31% at 40°C, to 54% at 60°C, whereas there was no significant effect of temperature on the reactivity of PS alone given in Fig. 2a. It has been reported that higher temperatures increase the production of
As given in Fig. 5, initial pH was adjusted at 3 (acidic), 7 (neutral), and 9 (basic) without using any buffer species that also react with radicals and cause analytical inhibition. PS and PMS in the presence of ZVI showed slightly better PFOS removal at acidic pH (Wang and Wang, 2018). PFOS removal by HP in the presence of ZVI greatly changed upon pH. Basic pH was beneficial to PFOS removal. Previous studies proposed that HP can be activated by Fe ions at basic pH to generate
Discussion on removal mechanism
Considerable removal of PFOS was observed in most of the cases tested. Because the integrated system uses solid ZVI, two possible mechanisms are considered to explain the observed PFOS removal: physical removal of PFOS to ZVI-associated species, and/or chemical decomposition of PFOS by oxidation and/or reduction. In all the cases including the case showing the highest PFOS removal in Fig. 3c for ZVI+HP at 60°C, increase in fluoride ion levels in the aqueous phase was not noticeable over the reaction time. Fluoride potentially on ZVI surface was also extracted, but fluoride concentration was negligible. Although formation of hydrofluoric acid could potentially occur under the final pH range of 1.5–4.5, we stopped further investigating fluorine species assuming no significant defluorination (no reaction byproducts were observed later). Thus, the PFOS removal can be explained by its adsorption to and complexation with Fe species such as ZVI, Fe oxides, and dissolved Fe ions and oxidation to unidentifiable intermediates (i.e., no significant defluorination of PFOS). To clarify the mechanism between adsorption and oxidation, F19-NMR and LC-MS analyses of the samples were introduced. NMR did not detect any significant alteration of PFOS structure. Furthermore, MRM mode was used in MS/MS analysis to search for targeted reaction intermediates, particularly shorter chain PFAS that were expected to be produced during the decomposition of PFOS. However, expected intermediates were not detected.
The results possibly propose adsorption as a major mechanism to explain the observed PFOS removal and raise many possible complex scenarios occurring in the ZVI-based integrated system. Previous studies reported that adsorption of PFAS onto ZVI surface was considerably high (Lawal and Choi, 2018; Park et al., 2018; Zhang et al., 2018). Although ZVI itself does not show significant adsorption of PFOS, corrosion layers of Fe oxides and Fe hydroxides formed as a result of ZVI oxidation might adsorb PFOS better (Kim et al., 2018). For example, adsorption of PFOS onto RNIP and ZVI alone with high surface area of >30 m2/g was negligible (Fig. 1b). As also given in Fig. 3, ZVI combined with any oxidants at 60°C showed higher PFOS removal, whereas ZVI alone at 60°C showed slight or negligible PFOS removal. This strongly suggests ZVI, when fast oxidized by added oxidants, can remove PFOS better.
The results given in Fig. 4 might also support the removal mechanism. Despite 10 times higher concentration of oxidants where much more radicals were expected to be generated, PFOS removal was only slightly improved. It should also be noted that excess oxidant is capable of scavenging generated radicals (Nfodzo and Choi, 2011). The PFOS removal cannot be explained by the radical generation mechanism. Rather, higher oxidant dose may facilitate corrosion of ZVI and thus cause fast adsorption of PFOS to newly formed oxide and hydroxide layers with the chemical affinity for PFOS (Choi et al., 2009b). Significant amounts of PFOS were removed in all the cases involving ZVI and oxidant. Based on the formation of Fe oxides around core ZVI particle because of its corrosion as observed through XRD analysis of spent ZVI and reported elsewhere (Choi et al., 2009b), the effect of solid Fe2O3 (Fe3+) and FeO (Fe2+) on the removal (presumably adsorption) of PFOS under different pH conditions was revealed in Fig. 6. Acidic pH compared with basic pH and Fe2O3 compared with FeO seemed more beneficial to PFOS removal.
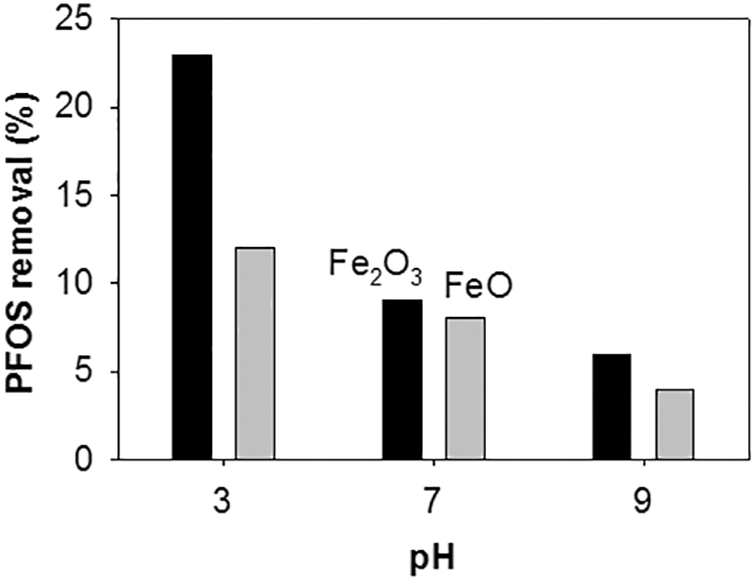
PFOS removal by adsorption to solid Fe2O3 and FeO particles under different initial pH conditions (10 mg/L PFOS; 0.5 g/L as Fe; initial pH 3, 7, or 9; 60°C). Fe, iron; Fe2O3, ferric oxide; FeO, ferrous oxide.
However, in the experiment conducted under pH 3 (e.g., in Figs. 5 and 6), Fe species were soluble under the pH condition and thus a small amount of solid ZVI and/or Fe oxides was available as potential PFOS adsorption sites. To further explain the high PFOS removal even in the case, PFOS removal was attempted with dissolved Fe3+ and Fe2+ ions, as given in Fig. 7, where Fe ions have the potential to bind PFOS to make a complex. Apparently, higher Fe doses and Fe3+ ions compared with Fe2+ (as the finding in Fig. 6) exhibited better PFOS removal. At concentration of 2 g/L of Fe ions under acidic conditions, PFOS removal by Fe3+ was at 20%, whereas that by Fe2+ was at 9%. Fe3+ showed better capability to make complex with PFOS. Similar results were reported by a study on the complexation of PFOS with different valence states of Fe (Park et al., 2018). Such Fe–PFOS complexes pose a challenge for prediction and quantification through MS analysis.

PFOS removal by complexation with dissolved Fe3+ and Fe2+ ions under different Fe doses (10 mg/L PFOS; 0.25–2 g/L as Fe; initial pH 3; 60°C). PFAS, polyfluoroalkyl substances.
The removal of PFOS through either adsorption or complexation (i.e., no chemical decomposition) posed a question about the PFAS treatment potential of the proposed strategy combining oxidation and reduction. We briefly checked if the complexity of the removal mechanism is only for PFOS or is it consistent to other PFAS. PFOA, with a very similar structure to PFOS (both are C8 PFAS), was quickly examined. Figure 8 shows removal of PFOA and PFOS in comparison by using ZVI+PS system. PFOA removal was comparable with PFOS removal. PFOS removal was faster during the initial phase and then removal of both PFOA and PFOS later was very similar. Compared with no identifiable intermediates from PFOS, targeted MS/MS analysis indicated formation of several reaction intermediates from PFOA, that is, shorter chain PFAS such as PFHpA (C7), PFHxA (C6), PFPeA (C5), and PFBA (C4), as given in Fig. 9. The mechanism of PFOA decomposition seems to be a progressive removal of CF2 moieties to form the shorter chain PFAS (Hori et al., 2005; Bruton and Sedlak, 2018).

Removal of PFOS (empty circle) and PFOA (solid circle) by ZVI conjugated with PS (10 mg/L PFOA or PFOS; 0.5 g/L ZVI; 0.3 M PS; initial pH 3; 60°C). First-order reaction model lines are also shown for PFOA (solid line) and PFOS (dotted line). Insets are the chemical structure of PFOS and PFOA with multiple C–F bonds. PFOA, perfluorooctanoic acid.

Identification of reaction intermediates formed during the decomposition of PFOA by ZVI conjugated with PS. Confirmation of the reaction intermediates was undertaken using targeted analysis based on our expectation, whereas many other intermediates were also formed.
Now, the observed decomposition of PFOA by the ZVI-based integrated system raises more questions that should be investigated in near future. PS itself is capable of decomposing PFOA in literature but it requires high temperatures (Bruton and Sedlak, 2018). In this study, PS is activated by Fe ions released from ZVI, along with heat activation of PS. The proposed ZVI-PS system at high temperatures might generate reactive species more effectively than only PS at high temperatures, resulting in potential reduction of temperature requirements for PFAS decomposition. Nonetheless, the ZVI-based integrated system combining oxidation and reduction works for the chemical decomposition of PFOA (and presumably other PFAS, in particular, carboxylic PFAS), but not for PFOS. Previous studies also reported a greater persistence of PFOS compared with PFOA presumably owing to the carboxylic functional group in PFOA being more vulnerable to chemical reaction than the sulfonic functional group in PFOS (Mitchell et al., 2014; Park et al., 2016; Bruton and Sedlak, 2018).
The strategy of using ZVI conjugated with oxidants was initially thought to be less dependent on the functional groups, considering both molecules possess overall very similar molecular structures. Of interest, one (PFOA) was decomposed, whereas the other (PFOS) was not decomposed. It should be investigated that the finding is applicable to other carboxylic PFAS in comparison with other sulfonate PFAS. Many other possibilities are also under consideration, including formation of insoluble (thus undetectable) PFOS dimer as a result of PFOS decomposition. Along with elucidating the decomposition mechanism of PFOA and (if any) PFOS, many other PFAS with different chain lengths and functional groups should be further examined with the ZVI-based system to answer the questions.
Combining advanced oxidation with chemical reduction by exploiting various oxidizing and reducing reactive species generated in the proposed system turned out to be not so effective for PFOS decomposition but to work for PFOA decomposition. Defluorination should be preceded by other chemical or photochemical processes somehow for the decomposition of PFOS. Finally, if proven successful in removing PFAS (at least carboxylic PFAS) through adsorption, complexation, and/or decomposition, the concept of this study can be directly applied with reactive barrier concept incorporating ZVI and oxidant injection to treat groundwater or heterogeneous media including soil and sediment as demonstrated for other halogenated chemicals (Choi et al., 2015). Meanwhile, ZVI can also be incorporated into the mesoporous structure of activated carbon (so-called reactive activated carbon) in which adsorption to activated carbon can mediate chemical decomposition of even hydrophobic PFAS (Choi et al., 2009a). It is also known that Fe oxide layer formed onto core ZVI and the porous structure of activated carbon prevent fast oxidation of ZVI (saving the reactivity of ZVI for long-term operation) (Choi et al., 2009b). These all also need to be investigated in near future.
Conclusions
This feasibility study examined if the ZVI-based integrated system exhibiting the capabilities of advanced oxidation, reductive dehalogenation, and possibly adsorption can remove and decompose PFOS under various conditions. In most of the experimental cases, a substantial amount of PFOS was removed. However, building up of identifiable expected intermediates and fluoride ions in water was negligible. The observed PFOS removal could be ascribed to presumably its adsorption to and complexation with Fe species such as Fe oxides and dissolved Fe ions originated from ZVI particles. The brief comparison between PFOS and PFOA raised more questions because PFOA (and presumably other PFAS) can be decomposed by the treatment system, generating obvious reaction intermediates such as shorter chain PFAS. The results indicated that PFOS, unlike PFOA, seems much more stable even in the presence of the strong reactive species produced under the tested experimental conditions. Many possible complex scenarios occurring in the integrated system were discussed. In-depth studies are needed in near future to narrow down the complex scenarios. Additional PFAS with different structures should be examined to obtain insight into their chemical reactivity with the integrated system. Finally, modifications of the treatment system are highly needed to further enhance the observed decomposition kinetic of PFOA and tackle the most challenging PFOS, such as use of other oxidants (e.g., ferrate) and surface-modified ZVI, addition of catalysts, intermittent addition of oxidants, and prevention of initial oxidation to facilitate reduction.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the Department of Defense through the Strategic Environmental Research and Development Program (ER18-1482), the Water Research Foundation (4877 (U2R16)), and the University of Texas at Arlington through Interdisciplinary Research Program.