
Abstract
Our studies showed that the living irradiated Saccharomyces cerevisiae has a good biosorption capacity for strontium and high tolerance to irradiation, but the mechanisms of this bioaccumulation are largely unknown. The mechanisms of S. cerevisiae to Sr2+ bioaccumulation was investigated using proteomics and metalloproteomics approaches. We previously identified 52 differentially expressed proteins (DEPs) by comparing the effect of Sr2+ adsorption on the proteome of S. cerevisiae. The selected DEPs were further analyzed using bioinformatics methods. A protein–protein interaction (PPI) network was constructed and analyzed using Search Tool for the Retrieval of Interacting Genes (STRING) and Cytoscape. Functional and signaling pathways of the identified DEPs with significant p-values were identified. We screened for the hub DEPs Ssa3p (SSA3), Kar2p (KAR2), Ssa4p (SSA4), Act1p (ACT1), Tubulin alpha chain (TUB1), Tubulin beta chain (TUB2), and ATP synthase subunit alpha (ATP1). The strontium-binding proteins of the living irradiated S. cerevisiae were purified by immobilized metal affinity chromatography and the cellular disposition and function of metals during the metabolism of S. cerevisiae were identified by liquid chromatography coupled with tandem mass spectrometry. Finally, 32 strontium-binding proteins were identified, and STRING was utilized to construct the PPI of the strontium-binding proteins and the selected hub DEPs, which were also analyzed using bioinformatics methods. Studies have shown that vacuolar transport, endosomal transport, and vesicle-mediated transport are critical to Sr2+ bioaccumulation, so we screened out that KAR2 is related to strontium detoxification, Sr2+ is transported into the cell through Rsn1p (RSN1), and ACT1 is related to strontium transport into the vacuole.
Introduction
The development and utilization of nuclear technology had brought a great deal of radioactive wastewater to humanity. How to treat radioactive waste has always been a global problem. 90Sr is a member of radionuclides in radioactive wastewater, which does enormous harm to the ecological environment and human health (Lee et al., 2005; Park et al., 2010; Du Bois et al., 2012).
Biosorption method, which has been studied for decades and favored by more researchers in the last 10 years, has shown a series of advantages such as high biosorption capacity, low production costs, and less secondary sludge (Farooq et al., 2010; Barot and Bagla, 2011; Wang et al., 2015). However, most studies focused on the dead cell to remove metal ions. Compared with dead cells, living cells showed a better removal efficiency since it exited the active bioaccumulation process. Moreover, to improve the adsorption capacity of the living biosorbent for radionuclides, many researchers modified the living biosorbent by genetic engineering to enhance the ability of active bioaccumulation. Appukuttan et al. (2011) found that the recombinant Deinococcus radiodurans cells encoding the phoN gene were able to precipitate ∼90% of uranium within 6 h. In both yeast and plants, Draexl (et al. 2013) found that Sec22p/SEC22 regulates cesium accumulation, which indicates that Sec22 proteins play a role in vacuole function. Heuck et al. (2010) employed an unbiased approach to identify candidate genes compromising Sr+ accumulation by screening a yeast knockout collection. Vacuolar transport, endosomal transport, and vesicle-mediated transport are the highest enriched categories among the identified genes (Heuck et al., 2010).
When a microorganism is undergoing metabolism, its ion channels, transport proteins, etc. be all working, and radionuclides enter the cell inside through these methods (Hansda et al., 2016). Studies showed that the intracellular bioaccumulation was related to proteins (Srinath et al., 2002). Some researchers have found that iron, copper, and manganese ions can be transported into Saccharomyces cerevisiae cells with the help of transmembrane transporter Ftr1p, Ctr1p, and Smf1p, respectively (Dancis et al., 1994; Eide, 1997; O'Halloran and Culotta, 2000; Culotta et al., 2005). In S. cerevisiae, the mitochondrial carrier family protein Pic2 imports copper into the matrix (Vest et al., 2016). The vacuole of yeast is able to sequester large amounts of calcium (Ca2+) and the yeast cells lacking Pmc1p localized to vacuole have cellular Ca2+ levels two- to three-fold lower than normal (Marchi et al., 1999; Qiu et al., 2019). Hence, proteomics data can help us study the biological processes caused by S. cerevisiae bioaccumulation of strontium. Nevertheless, a traditional proteomics approach may ignore metalloproteins that only change when they bind to metals, and their expression is almost unchanged when the metal is removed, or those have relatively low abundance in a proteome. At the same time, the interaction of the metal with proteins may determine the function of the protein (Goering, 1993). Knowledge of the intracellular high-affinity metal-binding proteins may be pivotal in understanding metal–protein interactions.
In this study, proteomics and metalloproteomic analyses of the living irradiated S. cerevisiae have been carried out. We previously identified 52 differentially expressed proteins (DEPs) by comparing the effect of Sr2+ adsorption on the proteome of S. cerevisiae. The selected DEPs were further analyzed using bioinformatics methods and got hub DEPs SSA3, KAR2, SSA4, ACT1, TUB2, TUB1, and ATP1, which were most significantly related to intracellular protein transmembrane transport. The strontium-binding proteins involved in strontium transport, storage, and utilization in the irradiated S. cerevisiae were isolated and identified by integrating the powerful tools of strontium-immobilized metal affinity chromatography (IMAC), sodium dodecyl sulfate polyacrylamide gel electrophoresis, and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) (Sun et al., 2011). The information obtained from the identification and functional analysis of these Sr2+-induced DEPs and strontium-binding proteins may improve our understanding of strontium bioaccumulation in S. cerevisiae. Therefore, combining the analysis of DEPs and strontium-binding proteins, the key proteins are KAR2, ACT1, and RSN1. Combining the analysis of DEPs and strontium-binding proteins, our results indicate that endoplasmic reticulum (ER) stress may be related to the mechanism of Sr2+ detoxification in cells, and then Sr2+ is transported into the cell through Rsn1p and enters the vacuole of yeast through endocytosis. It is speculated that ACT1, KAR2, and RSN1 proteins may be related to the bioaccumulation of Sr2+ in S. cerevisiae. Our results provided clues to the proteins that may be involved in the bioaccumulation of Sr2+ in S. cerevisiae. Then, we are expected to construct genetically engineered strains of higher removal rates of strontium ions in radioactive wastewater. The effective treatment of radioactive wastewater is of great significance.
Materials and Methods
Microorganism and materials
S. cerevisiae (CICC 30225) cells were purchased from the China Center of Industrial Culture Collection (CICC). Yeast cells were cultured in the liquid media consisting of glucose (50.0 g), yeast extract (0.5 g), Na2HPO4 (0.5 g), (NH4)2SO4 (1.0 g), urea (1 g), and 1,500 mL distilled water, at a pH 6.5–7.0. In our previous studies (Qiu et al., 2017), we had used the cyclic irradiation method for culturing an irradiated S. cerevisiae, Y-7. In this article, Y-7 will be used as living biosorbents for further researches. The strontium nitrate [Sr(NO3)2] was used to prepare the strontium solution. The pH was adjusted by HCl and NaOH. All chemicals were of analytical grade unless otherwise stated.
Analysis of DEPs
Gene ontology and kyoto encyclopedia of gene and genome pathway analysis of DEPs
Qiu collected the living irradiated S. cerevisiae exposed to 0, 40, 200, and 400 mg/L (strontium concentration), detected more than 500 protein spots in gels with different expression abundances, and compared the images of untreated and the metal sorbed S. cerevisiae samples (Supplementary Fig. S1), and a few unmatched but highly repeatable spots were found from repeated experiments. It was deemed as the selection criteria that the protein abundance ratio of each spot (treatment/control) was more than 2.0-fold or less than 0.5-fold, so a total of 52 DEPs were identified (Supplementary Table S1) (Qiu et al., 2019). Kyoto Encyclopedia of Gene and Genome (KEGG) is a database resource that can be used to understand advanced functions and biological system (Kanehisa, 2002). Gene ontology (GO) is a major bioinformatics tool for annotating genes and analyzing the biological processes of these genes (Ashburner et al., 2000). Database for Annotation, Visualization, and Integrated Discovery (DAVID, version 6.8) was used to do functional and pathway enrichment analyses on the DEPs.
Construction of interaction network
A Search Tool for the Retrieval of Interacting Genes (STRING, version 11.0) (Szklarczyk et al., 2010) was utilized to search for the interaction objects of 52 DEP products and a protein interaction network was constructed.
Hub protein screening
Studies on a biology network indicated that majority of biological networks obey scale-free (scale-free) network attributes (Jeong et al., 2000), which mean that a small number of nodes in the network had a large number of connections, most nodes had only a few connections, and these few nodes were the network critical node (hub) (Yu et al., 2004). By using the interaction network node and the scale-free nature of interaction in the protein network, the statistics node and its distribution were analyzed and the center of the net, the hub protein was found.
Analysis of network module
The module of the whole network obtained previously was decomposed and the function modules with modular nature, including the proteins identified through analysis, were found. Gene Ontology (GO) annotations were performed using Cytoscape (version 3.6.1) (Smoot et al., 2010), Mcode (version 1.5.1) (Rivera et al., 2010) (module parameters: Degree cutoff 2, K-core 2), and Bingo (version 3.0.3) (Maere et al., 2005) software based on the hypergeometric distribution and function enrichment threshold of adj. p < 0.05.
Biosorption experiment
The cell culture and biosorption experiment were carried out according to the experimental method of Qiu et al. (2017). The initial Sr(NO3)2 concentration of 0, 40, 200, and 400 mg/L at pH 7, respectively. All experiments were implemented in triplicate. To express the biosorption effect of metal ions by the yeast, the biosorption efficiency R (%) was advanced and calculated according to the following equation:
The biosorption quantity of strontium (capacity) q (mg/g) was calculated using the following equation:
q (mg/g) is the amount of strontium absorbed per gram of dry weight; C0 and Ce (mg/L) are the initial and equilibrium concentration of the metal solution, respectively; and Cm (g/L) is the absorbent dosage of S. cerevisiae.
Proteins extraction
S. cerevisiae cells grown in 100 mL liquid media to midlog phase were pelleted at 5,000 g for 20 min at 4°C. Each protein sample was ground to a powder with a mortar and pestle in liquid nitrogen, and then suspended in an extraction buffer (10% w/v trichloroacetic acid/acetone, 0.07% w/v β-mercaptoethanol, and 1 mM phenylmethanesulfonyl fluoride) at −20°C for 1 h. The vacuum-dried pellets were obtained after centrifugation and rinse, and then were dissolved in 800 mL of cell lysis solution (2 M thiourea, 7 M urea, 1 mM phenylmethanesulfonyl fluoride, 65 mM dithiothreitol, 4% w/v CHAPS, and 0.5% v/v biolytes) (Qiu et al., 2019). The lysate was centrifuged at 12,000 g at 4°C for 10 min, and the supernatant was ready for IMAC. Three biological replicates were used for each sample so that statistically meaningful results can be obtained. Protein concentrations were determined using the Bradford method (Bio-Rad, Hercules, CA) (Ma et al., 2012).
Separation and isolation of strontium-binding proteins based on strontium-IMAC
The Sr-column used in our experiments was prepared by displacing the Ni2+ from a commercial 5 mL of Ni-column (Invitrogen, Carlsbad, CA) with 0.05 M EDTA (twice, 15 mL each), followed by two washes with double-distilled water (15 mL each) and two washes with 0.5 M NaOH (15 mL each) to negatively charge the column. The column was saturated with Sr(NO3)2 (5 mg/mL in water). The column was then washed with three column volumes of double-distilled water to remove unbound Sr2+ and equilibrated with 0.02 M phosphate buffer (pH 7.8) with 0.5 M NaCl (Qian et al., 2000). A Ni-column without loading metal ions was used as a control in all experiments.
The protein extract was dialyzed against IMAC-binding buffer (20 mM phosphate buffer containing 0.5 M NaCl and 3 M urea, pH 7.8), filtered through a 0.45 mm cellulose membrane, and then loaded with strontium column preequilibrated with binding buffer. Unbound protein was washed away with 10 mL of binding buffer and 10 mL of binding buffer plus 0.005 M imidazole. Metal-binding proteins were eluted with a phosphate buffer containing 0.05 M Na EDTA. The experiment was repeated three times.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed on a 12% separating and 6% stacking gels to separate proteins. After electrophoresis, the gels were stained with Coomassie brilliant blue R-250 staining solution (0.2% Coomassie brilliant blue R-250 in 50% methanol and 10% glacial acetic acid) for 6 h and the same solution without dye for destaining.
After destaining, the Coomassie-stained protein bands were manually excised and is subjected to In-gel trypsin digestion.
Identification of strontium-binding protein
An in-gel digestion protocol was adapted as described by Wang (Chen et al., 2005). The samples were analyzed using a Capillary Liquid Chromatography–Electrospray Ionization–Quadrupole Time-of-Flight–Mass Spectrometry (CapLC-ESI-Q-TOF-MS). The automatic injection system for CapLC is equipped with a C18 desalination precolumn and a C18 capillary separation column. The experiment was carried out according to the method of Escobosa et al. (2015). The data after mass spectrometry are presented in the peak list file. These peak list files were then submitted to MASCOT (www.matrixscience.com) search engine to get corresponding protein identity. The reliability of the identified proteins was assessed by the molecular weight search (MOWSE) score, and results in a score >30 in this experiment were considered significant (individual ion scores >30 indicate the identity or extensive homology [p < 0.05]). GO analyses were conducted with the GO Slim Mapper from SGD (www.yeastgenome.org/cgi-bin/GO/goSlimMapper.pl).
Construction and analysis of the interactive network of strontium-binding proteins and screened differentially expressed proteins
STRING was utilized to search for the interaction objects of strontium-binding proteins and the screened DEPs, a protein interaction network was constructed, and then analyzed using Mcode and Bingo.
Results and Discussion
Analysis of DEPs
GO and KEGG pathway analysis of DEPs
As shown in Supplementary Table S1, 38 proteins showed upregulated expression profiles, 11 proteins showed downregulated and 3 proteins showed opposite expression profiles in different concentrations. GO analysis of the biological process, cellular components, and molecular functions and KEGG pathway analysis were carried out by the DAVID as shown in Table 1 and Fig. 1. As shown in the column height of Fig. 1, among all the DEPs, most enriched in the above three aspects of the proteins were: metabolic process, cytoplasm, and small molecule binding. KEGG pathway analysis revealed that the DEPs were mainly enriched in protein processing in the ER, Longevity regulating pathway—multiple species, Biosynthesis of amino acids, Biosynthesis of secondary metabolites, Phagosome, Arginine biosynthesis, and Carbon metabolism (Table 1). For annotations that may be related to the bioaccumulation of Sr2+ by S. cerevisiae, we screened one biological process and two biological pathways (Table 2) and screened out the key proteins: SSA3, KAR2, SSA4, ACT1, TUB2, and TUB1.
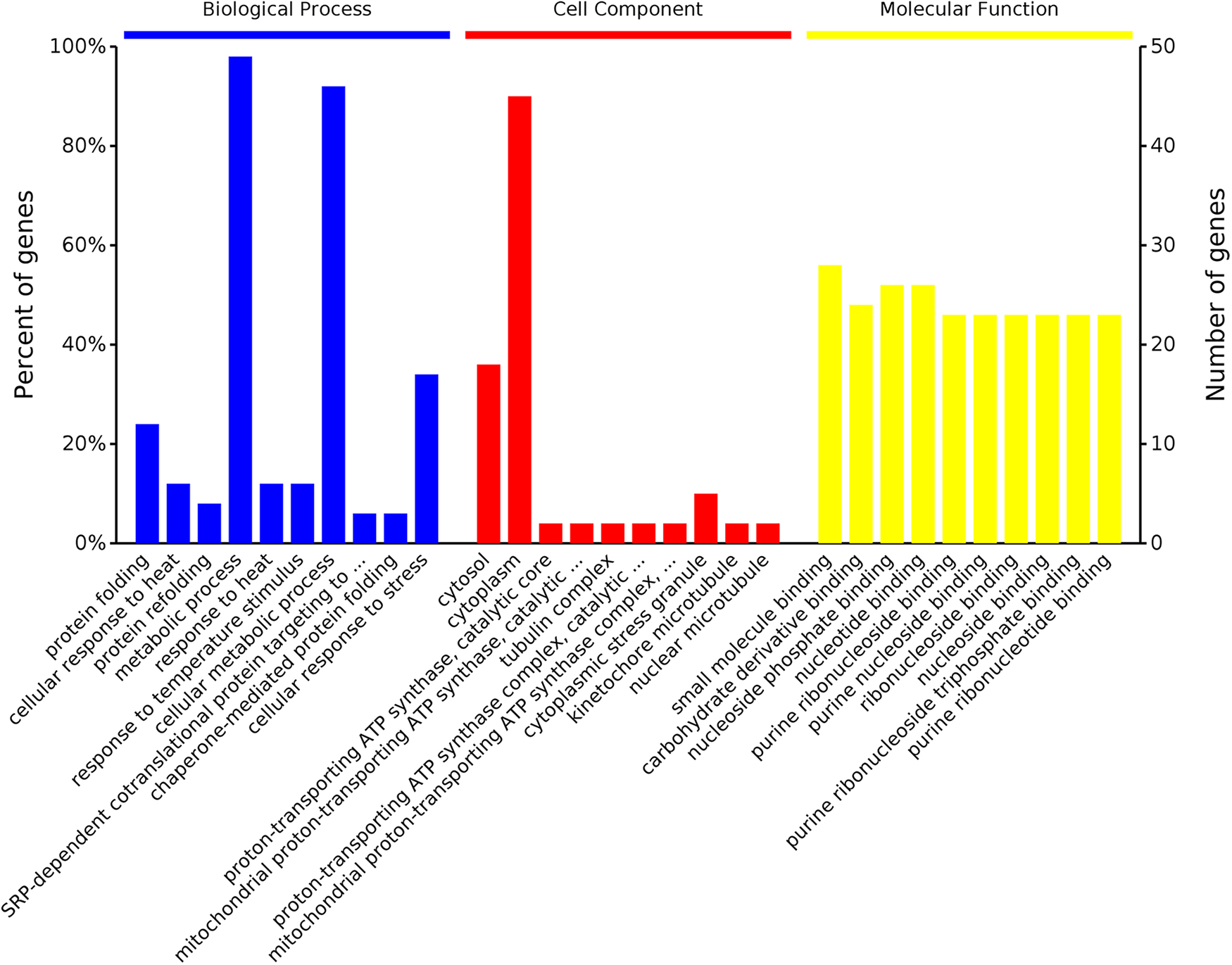
Enrichment pathways of DEPs in three aspects. From left to right were the biological processes, cellular components, and molecular functions. Abscissa axis represents the enriched function, the vertical axis represents the number of genes, and the height of the pillars represents the number of genes involved in the function. DEPs, differentially expressed proteins.
Gene Ontology and KEGG Pathway Enrichment Analysis of Differentially Expressed Proteins
ER, endoplasmic reticulum; GO, gene ontology.
Screening for Differentially Expressed Proteins of Interest
Screening for hub protein and the significant modular analyses through 52 differentially expressed protein–protein interaction network
Using the STRING software to search for protein–protein interactions (PPIs) of 52 DEPs, we obtained the protein interaction network, which consisted of 44 nodes connected through 175 edges as shown in Fig. 2A. We filtered 17 hub genes that were identified by filtering according to the criterion of degrees >10 criteria (each node had more than 10 interactions) as shown in Fig. 2A and Table 3. Statistical analysis of the node degree revealed that the highest node was the KAR2 protein (node 23), suggesting that these proteins play an important role in the network. The most significant module was obtained using Mcode, which is the plugin of Cytoscape, then garnished with another plugin Bingo to division and comments the whole network function module based on the hypergeometric distribution (adj. p < 0.05). It turns out that the most significant modules (Score: 10.364) involve proteins that are part of the above Hub proteins (Fig. 2B), and another seven notable features of interest were got from this module (Table 4). SRP-dependent cotranslational protein targeting to membrane, translocation functions, which contained SSA3, KAR2, and SSA4, was most significant in this module, in addition to the transmembrane transport functions, including SSA3, KAR2, SSA4, and ATP1.
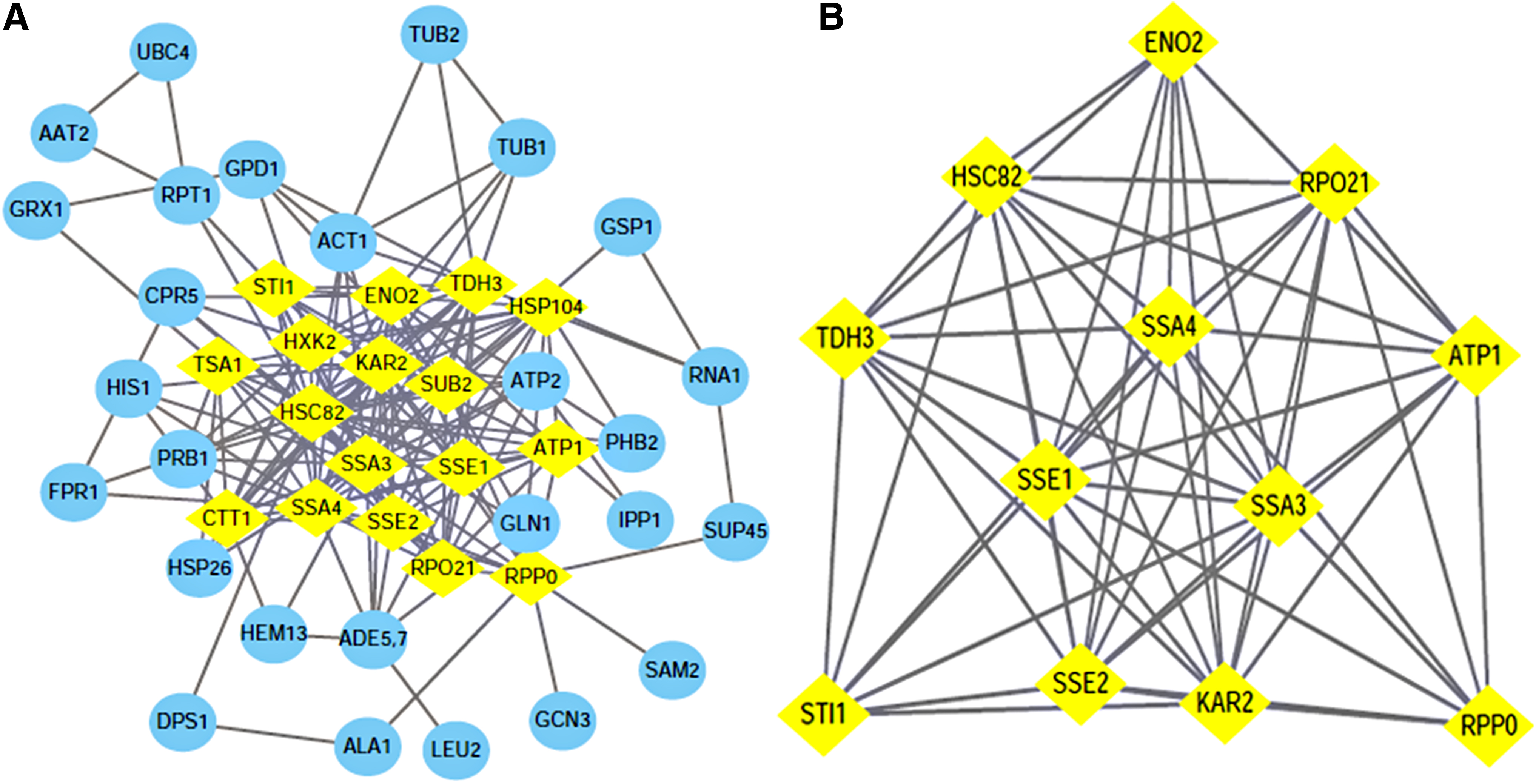
Node Degree of 17 Hub Proteins
Annotated List of Functions of the Most Significant Modules of Differentially Expressed Proteins
In summary, SSA3, KAR2, SSA4, ACT1, TUB2, TUB1, and ATP1 may be highly connected with the active bioaccumulation of strontium ions by the living irradiated S. cerevisiae.
Biosorption efficiency of the irradiated S. cerevisiae
The irradiated S. cerevisiae used in our study has high radiation resistance and stable gene inheritance (Tan et al., 2017). The biosorption efficiency (biosorption quantity) of the yeast in the environment of strontium with concentration of 40, 200, and 400 mg/L was 56.6% (11.14 mg/g), 85.7% (30.34 mg/g), and 67.3% (17.94 mg/g), respectively. The biosorption quantity of S. cerevisiae to Sr2+ at different strontium concentrations was consistent with our previous research results (Qiu et al., 2017).
Functional and localization analysis of strontium-binding protein
The electrophoresis results compared with the standard (marker) protein, protein band results are as shown in Fig. 3. The two bands of the sample are clear, and the results of the two parallel samples are basically the same, indicating the reliability of the experiment. It can be seen from Fig. 3 that the molecular weight of the strontium-binding protein is basically concentrated in the range of 55–70 kDa. Thirty-two proteins were isolated by strontium-IMAC. The detailed information about the identified proteins eluted from strontium-IMAC with their protein gene names is listed in Table 5. Since the control column was not retaining protein without metal loading, the proteins bound to strontium-IMAC were solely attributed to metal-binding abilities. The strontium-binding proteins were also not involved in the adsorption process, so the identified strontium-binding protein is not the same as the DEP. This indicates that metalloproteomics is an important complement to proteomics in the study of the biological mechanism of Sr2+ bioaccumulation. The proportion of proteins without annotation information is high, and we use BLAST to predict the function of these proteins.

SDS-PAGE gel of fractions eluted from Sr-IMAC (lanes 1–2). Lanes 1 and 2 (two parallel samples): fractions eluted with 0.05 M Na EDTA. IMAC, immobilized metal affinity chromatography; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; Sr, strontium.
Based on Strontium-Immobilized Metal Affinity Chromatography, 32 Strontium-Binding Proteins Were Identified in the Living Irradiated Saccharomyces cerevisiae
Accession number from the UniProt nr database.
Names of the genes obtained through the MASCOT software from the UniProt nr database.
Names of the proteins obtained through the MASCOT software from the UniProt nr database.
The sequence coverage of identified proteins.
MW and isoelectric point of the identified proteins.
MW, molecular weight; SC, sequence coverage.
The proteins in the identified S. cerevisiae were grouped into 13 manually curated, broad categories, based on the results of GO analyses conducted with the GO Slim mapper tool from the SGD database to give a general overview of all biological processes concerned as shown in Fig. 4.
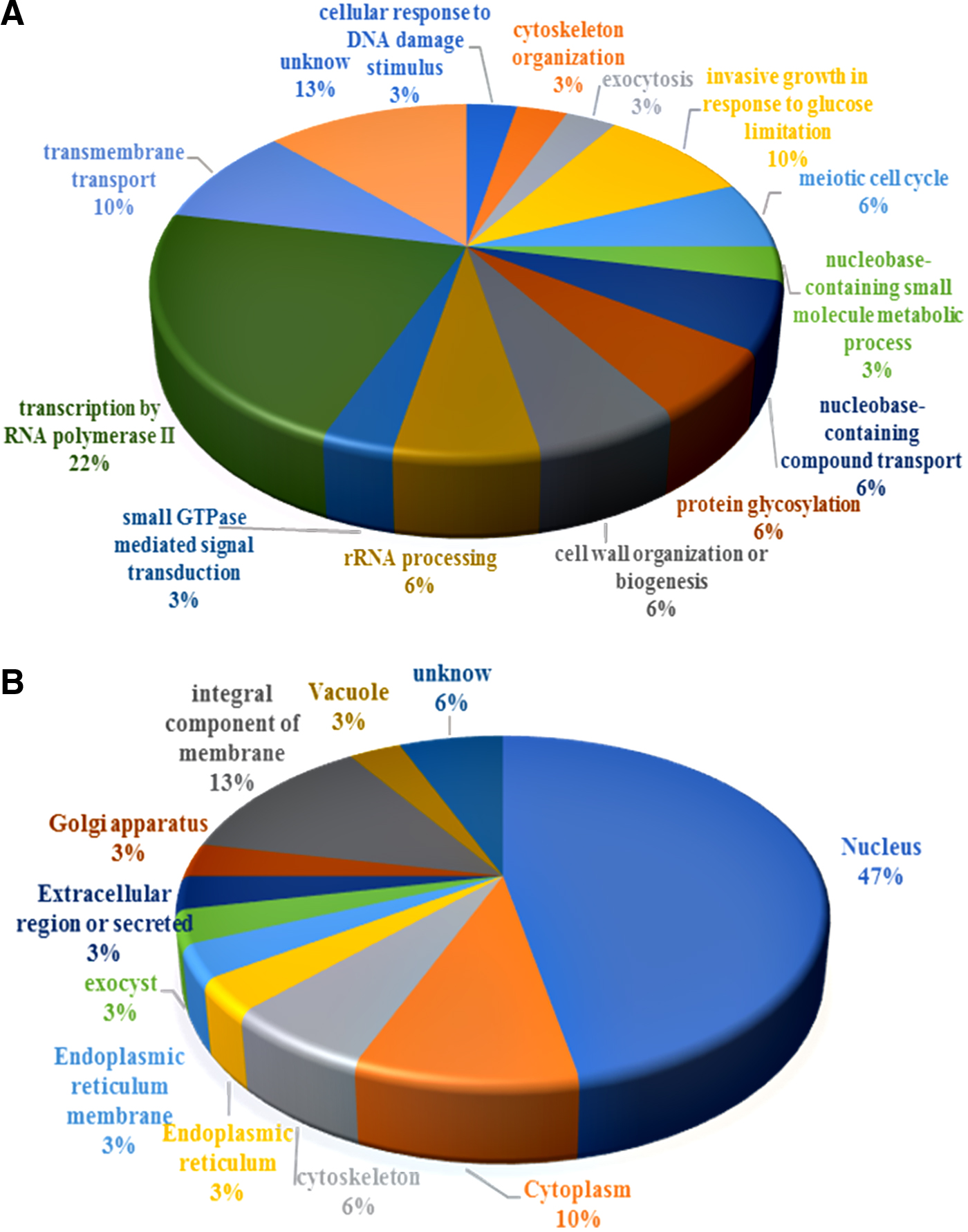
Functional classification of Sr-binding proteins in the irradiated Saccharomyces cerevisiae.
Construction and analysis of the interactive network of strontium-binding proteins and screened differentiall expressed proteins
Using the STRING software to search for PPIs of 7 DEPs and 32 strontium-binding proteins, we obtained the protein interaction network, which consisted of 21 nodes connected through 33 edges as shown in Fig. 5A. The most significant module was obtained using Mcode, which is the plugin of Cytoscape, then garnished with another plugin Bingo to division and comments the whole network function module based on the hypergeometric distribution (adj. p < 0.05). A module that contained the DEPs: SSA3, KAR2, SSA4, ACT1, TUB2 was obtained (Fig. 5B), and another five notable features were got from this module (Table 6).
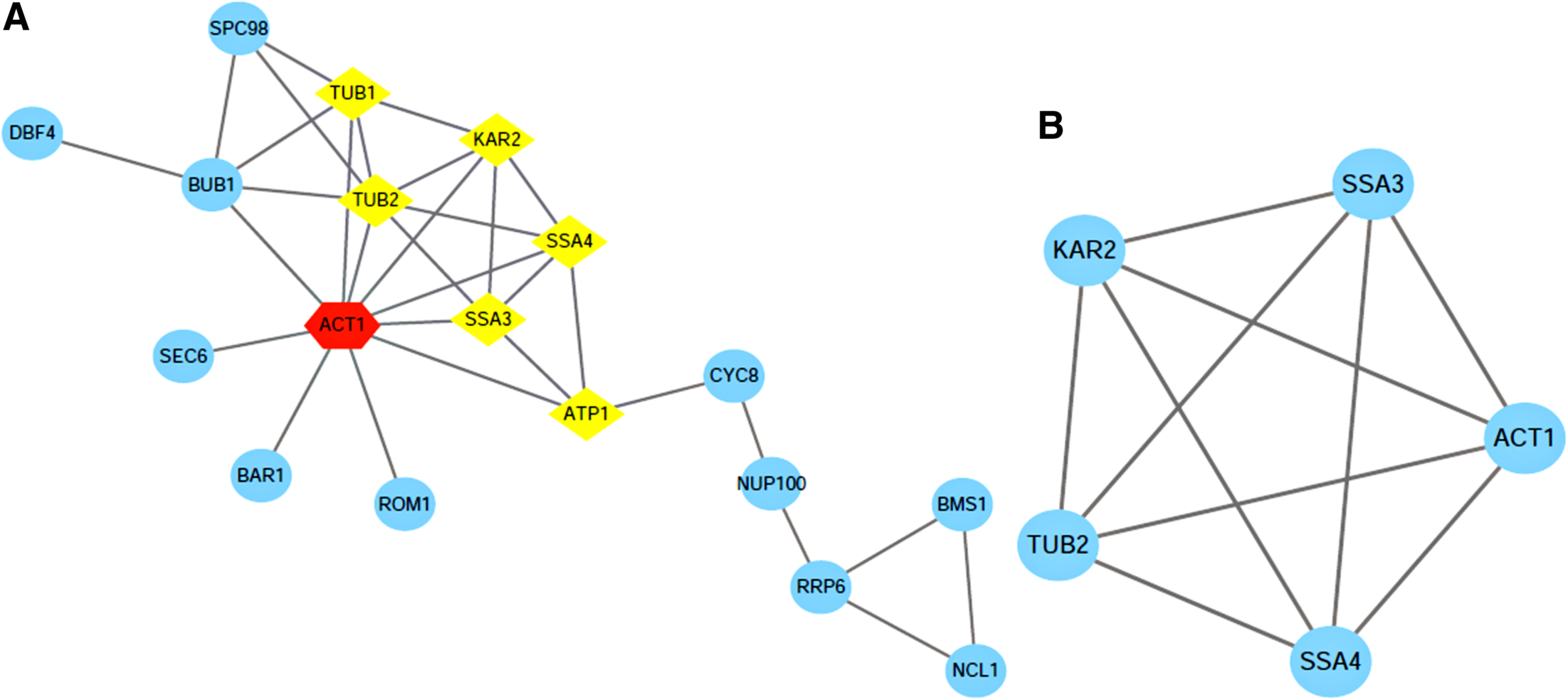
Annotated List of the Functions of the Most Significant Modules of 32 Strontium-Binding Proteins and 7 Differentially Expressed Proteins
Therefore, combining the analysis of DEPs and strontium-binding protein, the key research proteins are KAR2, SSA3, SSA4, ACT1, and TUB2. The previous differential expression results showed that KAR2, SSA3, TUB2, and ACT1 expressions were upregulated and SSA4 expression was downregulated.
Heat shock proteins (Hsps) were initially identified as expressing proteins induced by heat stress. In addition to playing a protective role under pressure, Hsp70 can also participate in transmembrane transport (Nollen and Morimoto, 2002; Sangster et al., 2004; De Los Rios et al., 2006; Floer et al., 2008). In S. cerevisiae, KAR2, SSA3, and SSA4 are all members of the Hsp70 family, of which SSA2 and SSA3 are localized in the cytoplasm or nucleus, and KAR2 are localized in the ER (Verghese et al., 2012). Besides, SSA3, SSA4, and KAR2 of the Hsp70 family have very high sequence similarity (Musso et al., 2007). KAR2, also known as Bip, is a homolog of the 78-kDa glucose regulatory protein (Grp78) and is the only Hsp70 chaperone in the ER, whose upregulation can relieve ER stress (Verghese et al., 2012).
The ER is the key organelle that controls protein folding, lipid biogenesis, and Ca2+ homeostasis (Bootman et al., 2002; Verkhratsky and Petersen, 2002). Due to the chemical similarity of strontium and Ca2+, the biological behavior of strontium is similar to that of Ca2+. Early experiments with various plants indicate that high Ca2+-accumulating species also tend to retain higher amounts of Sr2+ (Gregory, 2003). Comparing Ca2+ and Sr2+, classic kinetic studies in S. cerevisiae show that the uptake patterns of the two ions are highly similar and the affinity is comparable (Borst-Pauwels, 1981; Borst-Pauwels and Theuvenet, 1984).
In yeast cells, Ca2+ signaling mechanisms are regulated by the Cod1/Spf1 ATPase (Okorokova-Façanha et al., 2016) and Pmr1 Ca2+-ATPase (Antebi and Fink, 1992). Studies have shown that Cd exposure increases Ca2+ levels (Beyersmann and Hechtenberg, 1997) and Cd interacts with Ca2+ transport in intracellular stores, such as interference in hepatic Ca2+ sequestration in the microsomes (Zhang et al., 1990). KAR2 contributes to the buffering of Ca2+ in the ER lumen, and the irradiated S. cerevisiae shows high expression when adsorbing Sr2+ and yeast adsorbing cadmium, so both Sr2+ and Cd2+ may cause ER stress (Vido et al., 2001; Prins and Michalak, 2011). Studies have also shown that Cd2+-induced ER stress and Cd2+ toxicity are direct results of Cd2+ accumulation in the ER (Gardarin et al., 2010).
Vacuolar proton pump subunit B (VMA2) and RSN1 related to Ca2+ channels have also been identified in EDGs, as shown in Supplementary Table S1. VMA2 is proton-transporting V-type ATPase. V-ATPases acidify a series of endocytic compartments, terminating at the vacuole/lysosome, and also acidifying secretory vesicles. On the vacuolar membrane, the pH gradient generated by the V-ATPase activates the transporter, driving the uptake of ions and amino acids into the vacuolar lumen (Li and Kane, 2009). V-ATPase is mainly used as a driving force rather than a transporter to maintain intracellular Ca2+ levels (Forster and Kane, 2001).
Through the Uniport functional annotation, RSN1 is an osmotically sensitive Ca2+-permeable cation channel, located in ER, plasma membrane, and fungal-type vacuole membrane with the existence of several putative transmembrane domains in its sequence. It has been reported that overexpression of RSN1 inhibits the NaCl sensitivity of sro7 mutant cells by restoring the sodium pump (Ena1p) to the plasma membrane (Wadskog et al., 2006). The main function of the sodium pump is to remove excess Na+ from the cell and remove K+ from the cell into the membrane. Interestingly, we also identified a protein that has a high affinity with strontium, Protein HPH1 (FRT1), which is a calcineurin-dependent protein required for the growth of high NaCl, alkaline pH, and cell wall stress (Heath et al., 2004). We have done the effect of coexisting ions on the absorption of Sr2+ by irradiation of S. cerevisiae, and the results show that K+ promoted the adsorption of Sr2+ (unreported), so it is speculated that K+, Sr2+, and Ca2+ transport mechanisms are related. Our results indicate that the expression of this protein corresponds to the adsorption capacity of strontium (1.538, 2.111, and 1.759).
Among the identified DEPs, the highest expression abundance is Phosphotransferase (HXK2; 7.2036, 7.8936, 9.8633), and the second is ACT1 (5.3761, 8.3137, 1.7547). HXK2 protein is associated with glycolysis, and the glycolysis pathway may be necessary for metal-induced apoptosis, and studies have shown that S. cerevisiae undergoes a glucose-dependent, programmed cell death in response to low cadmium concentrations (Nargund et al., 2008). It has already been shown that the genes and proteins associated with glycolysis were overexpressed under acetic acid stress (Almeida et al., 2009; Mira et al., 2010) in response to the demand for ATP (Almeida et al., 2009; Cacho Teixeira et al., 2009). In addition, cells lacking HXK2 are sensitive to acetic acid, which is an indicator of programmed cell death, so HXK2 plays an important role in the apoptosis pathway (Konarzewska et al., 2017). Therefore, in our experiments, we speculated that HXK2 may be related to strontium-induced apoptosis. Interestingly, ACT1 was identified in both DEPs and strontium-binding proteins, so it is not only related to the bioaccumulation of the irradiated S. cerevisiae, but also has a high affinity for strontium.
ACT1 is actin, a structural protein involved in cell polarization, endocytosis, and other cytoskeletal functions. Endocytosis, a process that internalizes solutes, receptors, and plasma membrane transporters through endocytic vesicles and early and late endosomes, thereby delivering the cargo to the vacuole (Rotin et al., 2000). In our previous study, we used transmission electron microscopy and energy dispersive X-ray spectrometry to observe the intracellular structure of the irradiated S. cerevisiae after biosorption of Sr2+, and found that the vacuole was destroyed (Qiu et al., 2018). Heuck et al.'s (2010) study has shown that the destruction of the vacuole will enhance the bioaccumulation of Sr2+. This may be due to the fragmentation of the vacuole caused by the osmotic reaction increasing the surface area/volume ratio of the vacuole, which may allow the transporter to sequester cations into the vacuole to the greatest extent (Liu et al., 2005; Breuer et al., 2008; Dove et al., 2009; Li and Kane, 2009). Therefore, Sr2+ may be transported into the vacuole through endocytosis.
Endocytosis requires a properly functioning actin skeleton, which is a dynamic structure that suggests that actin may play a role in membrane invagination, vesicle contraction, vesicle scission, and vesicle movement (Geli and Riezman, 1998). These vesicles can be classified by their different proteinaceous coats into COPII, COPI, and clathrin-coated vesicles. At the ER–Golgi interface, newly synthesized proteins are transported from the ER to the Golgi apparatus in COPII vesicles, while COPI vesicles transport cargo, such as escaped ER-resident proteins, from the Golgi apparatus back to the ER. Studies have shown that KAR2/BiP is a soluble ER protein, which is recovered from the Golgi after escaping from ER (Semenza et al., 1990). Therefore, in our experiments, we speculate that Sr2+-induced ER stress is related to endocytosis, which may also affect the bioaccumulation of Sr2+.
TUB2 associates with alpha-tubulin (TUB1 and TUB3) to form tubulin dimer, which polymerizes to form microtubules. Rab GTPases have been shown to be implicated in the regulation of almost all steps in membrane transport, including vesicle formation (Morsomme and Riezman, 2002). In higher eukaryotes, transport along microtubules plays an important role in vesicular transport, for example, in the transport of synaptic vesicles, and besides, it has been shown to be the Rab effect (de Hoop et al., 1994; Grosshans et al., 2006).
Therefore, we hypothesized that Sr2+ induces ER stress, and ER stress is related to the mechanism of Sr2+ detoxification in the cell, and then Sr2+ is transported into the cell through RSN1 and enters the vacuole of yeast through endocytosis. Thus, it is speculated that ACT1, KAR2, and RSN1 proteins might be related to the bioaccumulation of Sr2+ by S. cerevisiae.
Conclusion
In this article, 52 DEPs identified previously were analyzed by Bioinformatics, and Hub proteins, SSA3, KAR2, SSA4, ACT1, TUB2, TUB1, and ATP1, were obtained. By studying strontium-binding proteins, strontium ions may be bound to proteins after bioaccumulation. Combined with the identified 32 strontium-binding proteins and literature searching, the key proteins ACT1, KAR2, and RSN1 related to the bioaccumulation of Sr2+ were obtained.
Increasing or decreasing the expression of the two proteins by the way of genetic engineering can confirm whether the two proteins are related to the intracellular bioaccumulation of strontium. We have paved the way for further research on the intracellular bioaccumulation mechanisms of strontium by the irradiated S. cerevisiae.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.