
Abstract
Manganese oxide (MnO2) amendments to mercury (Hg)-contaminated sediment have been shown to decrease methylmercury (MeHg) concentrations in sediment porewater. In the absence of solid-phase MeHg measurements, it is unclear whether the decreased porewater concentrations are due to inhibition of methylation, adsorption of the MeHg to the MnO2, demethylation of MeHg catalyzed by the MnO2, or some combination of these mechanisms. We conducted controlled laboratory experiments to determine whether MeHg losses from solution in the presence of amorphous MnO2 are due to sorption or demethylation. We quantified MeHg in the dissolved phase and solid phase and found that no demethylation occurred. We used batch and isotherm experiments to determine MeHg sorption efficiency to MnO2 in a variety of solutions with varying ionic strength and dissolved organic matter (DOM) concentration. Isotherms over an initial concentration range of 5–500 ng/L of MeHg showed no saturation effects. Increasing ionic strength from 0.012 M to 0.1 M produced relatively minor decreases in MeHg sorption. Increasing DOM concentrations from 0.64 to 16.4 mg C/L yielded substantial decreases in MeHg sorption. Under the experimental conditions, MnO2 is a less efficient MeHg sorbent compared to other sorbent materials, such as Thiol-SAMMS®, activated carbon, and biochar.
Introduction
Methylmercury (MeHg) is a known reproductive hazard and neurotoxin present in the environment (Clarkson et al., 2003; Hong et al., 2012). MeHg is formed when inorganic mercury (Hg) is methylated in a process mediated by a diverse group of anaerobic microorganisms, including sulfate-reducing, iron-reducing, and methanogenic bacteria (Gilmour et al., 2013a; Bravo and Cosio, 2019). The primary zones of MeHg production are found in anoxic aquatic environments, such as anoxic sediments (Hsu-Kim et al., 2018; Bravo and Cosio, 2019). MeHg bioaccumulates in benthic organisms and biomagnifies up the food chain posing a risk to top consumers (Hong et al., 2012; Moyo, 2020). The most common exposure route for humans is fish consumption (Clarkson et al., 2003; Hong et al., 2012). While MeHg formation in the environment is a biologically mediated process, MeHg demethylation, the process by which MeHg is broken down into inorganic Hg, can be mediated by both biotic and abiotic mechanisms (Pak and Bartha, 1998; Marvin-DiPasquale et al., 2000; Barkay et al., 2003; Qian et al., 2014). Microbially mediated demethylation can yield either methane or carbon dioxide from the methyl moiety of MeHg.
Dredging and landfilling MeHg-contaminated sediment have been a common remediation approach as has in situ capping, but these methods can be costly and ecologically destructive (Randall and Chattopadhyay, 2013; Hsu-Kim et al., 2018; Eckley et al., 2020). In situ treatments have also been studied, with much of the attention given to adsorbents (Johs et al., 2019; Muller et al., 2019; Eckley et al., 2020). Sorbents blended into sediments or applied as a permeable capping layer may be less ecologically disruptive than excavation or hydraulic isolation capping. Engineered sorbents (e.g., biochar, SediMite™ and Thiol-SAMMS®) are designed to remove inorganic Hg from the aqueous phase, potentially preventing its methylation (Gomez-Eyles et al., 2013; Johs et al., 2019), and may remove MeHg from the aqueous phase, potentially decreasing its bioaccumulation (Gomez-Eyles et al., 2013; Muller et al., 2019).
Sorbent amendments, such as activated carbon (AC) and biochar, have been used in laboratory experiments (Gilmour et al., 2013b; Lewis et al., 2016; Liu et al., 2018; Ting et al., 2018; Schwartz et al., 2019) and experimental field plots (Lewis et al., 2016; Gilmour et al., 2018) to decrease MeHg concentrations in sediment porewater. Porewater concentrations are a commonly used predictor for MeHg bioaccumulation. In aqueous solutions, sulfidized materials (e.g., Thiol-SAMMS) consistently show the highest sorption efficiency (Muller et al., 2019; Eckley et al., 2020). AC, biochar, and organoclay also decrease dissolved MeHg concentrations (Gomez-Eyles et al., 2013; Liu et al., 2018; Ting et al., 2018; Muller et al., 2019; Eckley et al., 2020), with AC showing the most consistent and effective performance. When efficiency, cost, and availability are considered, AC-based SediMite performed better for MeHg removal than several other engineered sorbents (Muller et al., 2019).
Although sorbents may work effectively for MeHg in simple, aqueous solutions, various conditions present in the natural environment can decrease sorption. Dissolved organic matter (DOM) is ubiquitous in the environment and can decrease sorption onto amendments through formation of stable aqueous complexes with lower affinity for sorbent surfaces (Johs et al., 2019; Muller et al., 2019; Schwartz et al., 2019). Hg-DOM and MeHg-DOM complexes appear to have a lower affinity for sorbents, with their sorption behavior more closely mirroring the sorption of DOM than Hg (Johs et al., 2019; Muller et al., 2019; Schwartz et al., 2019). DOM may also lower sorption by competing for sorption sites with the sorbate-DOM complex (Johs et al., 2019; Muller et al., 2019). Ionic strength reduces sorption of MeHg and Hg onto minerals, such as kaolin (de Diego et al., 2001), but we are not aware of any studies explicitly examining the effect of ionic strength on MeHg sorption onto AC or other sorbent amendments. In the case of Hg, increasing salinity or ionic strength results in minimal to moderate decreases in Hg sorption to sorbent materials, possibly due to changes in the Hg speciation that affect the sorption affinity of Hg (Wang et al., 2009; Boutsika et al., 2017). However, these studies were conducted in mg/L Hg solutions, which are higher than concentrations encountered at even the most contaminated sites.
Manganese (Mn) oxides have been proposed as a potential in situ amendment for mitigating Hg and MeHg contamination (Vlassopoulos et al., 2018; Eckley et al., 2020). Multiple studies have shown that Mn oxides can have an inhibitory effect on MeHg production (Jackson, 1989; Farrell et al., 1998; Vlassopoulos et al., 2018). MnO2 (in the form of pyrolusite or birnessite) decreased MeHg concentrations (66–89%, depending on MnO2 type) in sediment porewater when the MnO2 was mixed with contaminated sediment or applied as a thin-layer amendment to the top of the sediment (Vlassopoulos et al., 2018). Mn-reduction was confirmed as the dominant redox process in the mesocosms, indicating that the MnO2 may have poised the redox potential above conditions that are conducive to Hg-methylation (e.g., Fe(III)- and sulfate-reducing conditions). However, the solid phase was not analyzed so sorption or abiotic MeHg demethylation by MnO2 could not be ruled out as mechanisms contributing to lower porewater MeHg. Mn oxide-coated clay inhibited microbial activity and the associated microbially mediated MeHg production under certain nutrient conditions (Jackson, 1989). In the absence of microbes, MeHg was rapidly lost from solution in the presence of Mn oxide-coated clay, possibly due to demethylation by the Mn oxide (Jackson, 1989). The mineralogy of the Mn-oxides synthesized in that study was not determined.
The objective of our study was to quantify MeHg losses from solution in the presence of MnO2 and further determine if the losses were attributed to sorption, demethylation, or a combination of these mechanisms. Particular care was devoted to verifying analytical methods and MeHg mass balance to account for MeHg losses from the aqueous phase of MnO2/MeHg mixtures. MeHg loss from solution was due to sorption, not demethylation. We used batch and isotherm experiments to investigate the partitioning of MeHg onto MnO2. Experiments were conducted in solutions with a range of ionic strengths, DOM concentrations, and DOM types (e.g., Suwannee River Natural Organic Matter [SRNOM] vs. natural creek water) to determine how these environmental parameters influence MeHg sorption to MnO2.
Materials and Methods
MnO2 synthesis
MnO2 was synthesized by adding concentrated HCl to 0.4 M KMnO4 at a ratio of 0.0654:1 (vol:vol) (Buser et al., 1954; Fendorf and Zasoski, 1992). The resulting solid was washed with distilled, deionized water, dried at 60°C, and ground to a fine powder. The final product had a specific surface area of 28.35 m2/g and an average Mn oxidation state of 3.93 ± 0.02 as determined by oxalate reduction-permanganate titration. Random powder mounts showed the MnO2 solids to be amorphous by X-ray diffraction analysis.
Fate of MeHg in contact with MnO2
Batch experiments were run to track the partitioning of MeHg from the aqueous phase to the solid phase and to quantify any loss of total MeHg. A summary of experiments is provided in Table 1 with more detail provided below.
Summary of Experimental Conditions
pH increased 0.12 U over 120 h when mixed with the MnO2.
For the isotherm conducted in creek water, MeHg was added as Me202Hg to distinguish the added MeHg from ambient MeHg in the water.
PB, phosphate buffer.
General methods
All experiments were conducted in trace clean 40 mL amber glass vials (Thermo-Fisher). Unless indicated otherwise, 15–20 mL of air-equilibrated 5 mM phosphate buffer (PB; 1.038 g/L KH2PO4 and 0.1558 g/L K2HPO4, adjusted to pH 7.4 with NaOH) and 0.1 ± 0.01 g MnO2 were used as the matrix solution and solids added, respectively. The PB pH increased less than 0.13 units over 5 days when in contact with the MnO2. Other buffer systems created problems with the MeHg analysis, for example, precipitates formed when samples were acidified when using PIPES buffer. MeHg spikes were diluted from a MeHgCl stock standard purchased from Brooks Rand (Seattle, WA). Vials were protected from light and placed on a reciprocating shaker (Eberbach Scientific Instruments and Apparatus, Model 6010) on the low setting for up to 120 h at room temperature (23°C). The samples were then taken off the shaker, the aqueous phase decanted into a syringe barrel, filtered (0.2 μm Polyethersulfone (PES) syringe filter), preserved with trace metal-grade HCl (final concentration 0.5% v/v), and stored at 4°C until analysis. The residual solids were stored in the original vial at −20°C until further analysis. Three to six replicate vials were established per treatment. Controls without MnO2 included filtered and unfiltered MeHg solutions that were carried through the experiment procedure.
MeHg mass recovery
We assessed MeHg mass recovery from controls in which no MnO2 was present. MeHg recovery from unfiltered control samples in PB averaged 96% ± 6% (n = 13). In contrast, MeHg recovery from filtered control samples averaged 83% ± 7% (n = 24), indicating ∼15% of the MeHg was lost to the PES filter or its housing. This loss was independent of initial MeHg concentration or the presence of SRNOM. Therefore, in our data analysis, 15% was added to the aqueous phase MeHg results.
MeHg mass recovery was also assessed in PB + MnO2 slurry samples. The slurry samples were prepared as described above, and after 24 h, the whole sample was preserved in its vial with HCl and held at 4°C until analysis. The whole slurry sample was distilled and analyzed for MeHg.
Effect of ionic strength and DOM
The effects of solution ionic strength and DOM on MeHg-MnO2 interactions were studied at a single MeHg concentration of 102 ng/L. Other experimental conditions and procedures were the same as described earlier. The PB had an ionic strength of 0.012 M and ionic strength was adjusted to 0.05 M or 0.1 M with NaNO3. NaNO3 was used to adjust ionic strength to minimize potential confounding aqueous speciation effects that would have occurred using NaCl to adjust ionic strength (Supplementary Table S1).
DOM solutions were made by dissolving unfractionated Suwannee River natural organic matter (SRNOM; 2R101N from the International Humic Substances Society; 50.7% carbon, 3.97% hydrogen, 41.48% oxygen, 1.27% nitrogen, and 1.78% sulfur by weight) in PB. The effect of SRNOM on MeHg-MnO2 interactions was studied by preparing solutions of SRNOM (0.64, 2, and 16.4 mg C/L) in PB. These concentrations bracket typical dissolved organic carbon (DOC) concentrations (geometric mean and 95% CI = 2.16 [1.29–3.61] mg C/L, n = 164) at our sampling location in East Fork Poplar Creek (EFPC), an Hg- and MeHg-contaminated creek in Oak Ridge, TN (Brooks and Southworth, 2011).
Sorption Isotherms
Isotherm experiments were conducted using 5 mM PB, and 5 mM PB with 2 mg C/L SRNOM over a range of initial MeHg concentrations from 5 to 500 ng/L (Table 1). An additional isotherm was conducted using filtered (0.2 μm PES filter) EFPC surface water. Subsamples of the EFPC water were collected and preserved appropriately for additional ambient water quality characterization (Supplementary Table S2). To distinguish the added MeHg from ambient MeHg in EFPC (0.2 ng/L, Supplementary Table S2), isotopically labeled Me202HgCl was synthesized in-house using the methylcobalamin method (Bancon-Montigny et al., 2004) and was used to achieve final concentrations ranging from 5 to 500 ng/L.
Isotherm samples were equilibrated for 24 (PB and EFPC) to 72 (EFPC) h before sampling and preservation. Sorbed MeHg was directly measured by distilling the solids as described below (i.e., was not determined indirectly by the difference between initial and final aqueous concentration). The remaining experimental conditions and procedures were the same as described earlier.
Analytical methods
Aqueous and solid samples were analyzed for MeHg following standard methods (U. S. EPA, 2001) using Isotope Dilution-Gas Chromatography-Inductively Coupled Plasma-Mass Spectrometry (ID-GC-ICP-MS). Both filtrate and solid samples were distilled before analysis. A summary of average isotope dilution recovery and detection limit is given in Supplementary Table S3.
DOC was analyzed using a Shimadzu TOC-L analyzer. Anions in EFPC water were analyzed on a Dionex ICS-2100 with an IonPac AS9-HC column.
Isotherm modeling
Linear and Freundlich isotherms were fit to the data. The Langmuir isotherm was not considered because the data did not show saturation-type behavior. These isotherms take the following form:
Where S = amount sorbed (ng/kg); Ceq = equilibrium MeHg concentration in solution (ng/L); KD = linear partition coefficient (L/kg); and KF (ng1–n Ln/kg) and n (–) are Freundlich isotherm parameters. Each isotherm was fit to the directly measured aqueous- and solid-phase MeHg concentration data by adjusting isotherm parameters to minimize the sum of absolute relative errors (SARE), defined as
Where
Aqueous speciation modeling
The initial MeHg aqueous speciation was estimated using PHREEQC (Parkhurst and Appelo, 2013) and a database amended to include MeHg and its aqueous complexes (Supplementary Table S4). MeHg-DOM complexation was modeled assuming these interactions would be dominated by reduced sulfur (S) groups in DOM (RS–; MeHgRS) (Dong et al., 2010). The RS content of SRNOM 2R101N was estimated using the elemental composition analysis and equations provided in Dong et al. (2010) to be 5.475 × 10–4 millimole RS per milligram carbon. This model assumes all the RS is accessible to and has equal affinity for MeHg. In the PB matrix, in the absence of SRNOM, virtually all the MeHg was predicted to be present as either MeHgHPO4– or MeHgOH0 (Supplementary Table S5). In the presence of SRNOM or in EFPC, predicted initial MeHg speciation was dominated by MeHg-DOM complexes and was >98% MeHgRS for all DOC concentrations considered. In addition, speciation changes are minimal in the pH range of experiments, particularly in the presence of DOM (Supplementary Fig. S1).
Results and Discussion
MeHg mass recovery
When 102 ng/L MeHg was reacted with 0.1 g MnO2 in 15 mL PB, 36% ± 1% of the MeHg was recovered in the aqueous phase. When the whole slurry was digested, MeHg mass balance averaged 104% ± 5%. Similarly, MeHg mass balance for control samples averaged 100% ± 3%. These results indicate that under these conditions, no overall loss of MeHg occurred and that the loss of MeHg from the dissolved phase was due to sorption and not demethylation.
Ionic strength and SRNOM effects on MeHg partitioning to MnO2
Having determined that MeHg loss from the aqueous phase was due to sorption rather than demethylation, we conducted a series of experiments at a single MeHg concentration to explore the effects of ionic strength and SRNOM on MeHg sorption (Table 1). The results are compared by calculating a single-point linear partition coefficient [KD; Eq. (1)].
Effect of ionic strength
The base PB had ionic strength (IS) 0.012 M. To explore the effect of ionic strength on MeHg sorption, the PB was amended with either 40 or 90 mM NaNO3 to reach ionic strengths of ∼0.05 and 0.1 M, respectively. At the higher ionic strengths, the aqueous MeHg speciation changed from equal amounts MeHgHPO4− and MeHgOH0 at I = 0.012 to 62% and 60% MeHgOH0 at ionic strengths of 0.05 and 0.1 M, respectively, with the balance being MeHgHPO4− (Supplementary Table S5). Increasing the IS from 0.012 M to 0.05 M decreased the KD value from 320 ± 50 L/kg (log10 KD = 2.51 ± 0.07) to 228 ± 51 L/kg (2.36 ± 0.1), a 30% decrease (Fig. 1A). Further increasing IS to 0.1 M further decreased the KD to 195 ± 15 L/kg (2.29 ± 0.03). These relatively minor effects of IS on MeHg sorption suggest the mode of MeHg sorption remained similar over this range of ionic strengths. Apparent ionic strength effects may vary, depending on the strength of MeHg complexation with the electrolyte. In our study, we chose NaNO3 as an ionic strength adjustor to avoid confounding the effects of ionic strength and complexation affinity (Supplementary Table S1).
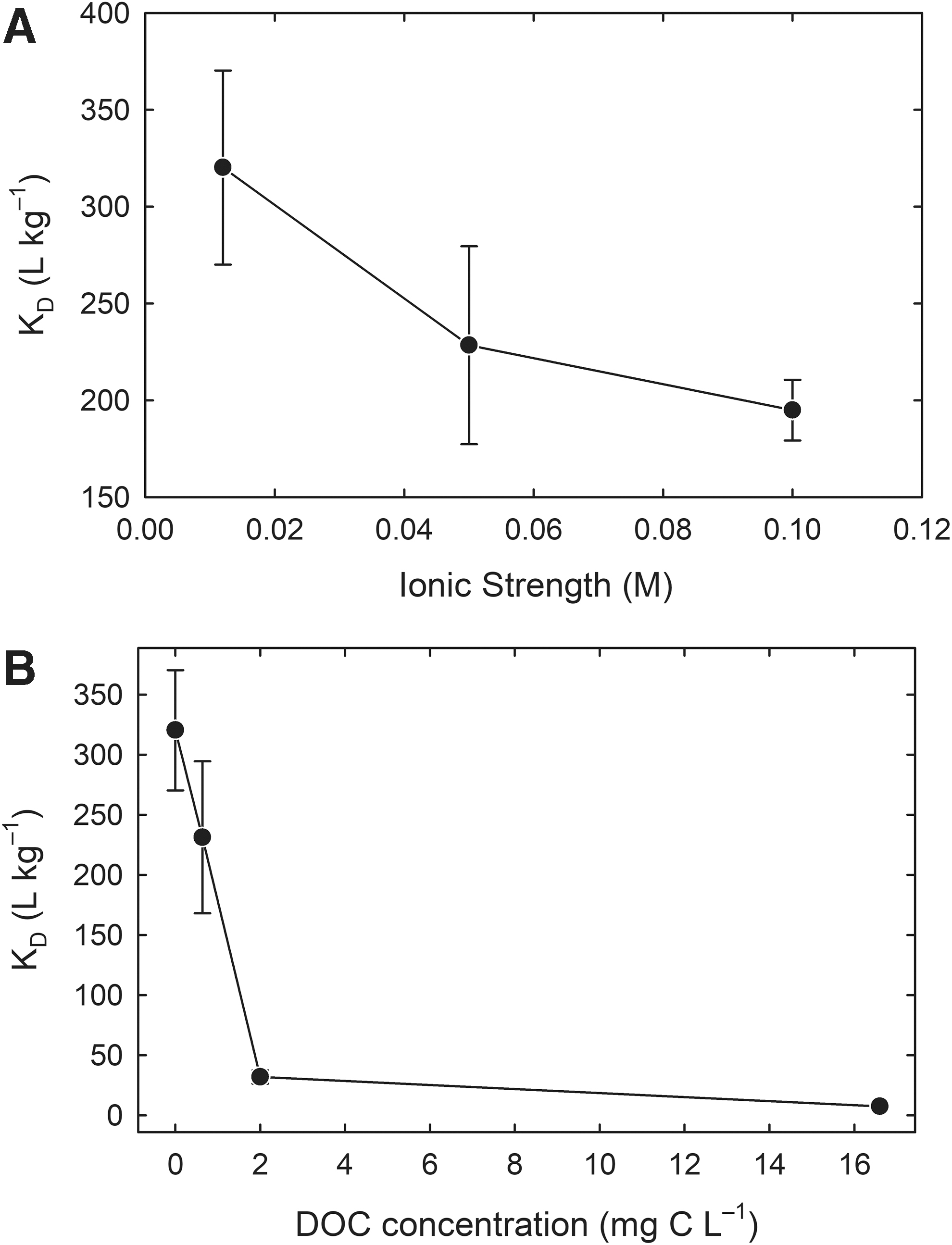
MeHg-MnO2 distribution coefficient as a function of
Effect of SRNOM
While ionic strength experiments are valuable in that they help elucidate the sorption mechanism of MeHg, in oxic freshwater systems, MeHg speciation is dominated by DOM complexes regardless of the ionic strength and electrolyte identity of the system (Hsu-Kim et al., 2013). To explore the effect of DOM on MeHg sorption, SRNOM was added at concentrations of 0.64, 2, and 16.6 mg C/L. These concentrations bracket typical baseflow DOC concentrations in EFPC. Predicted initial MeHg aqueous speciation was >98% MeHg-DOM across these SRNOM concentrations (Supplementary Table S5).
The MeHg partition coefficient decreased with increasing amounts of SRNOM from 320 ± 50 L/kg (log10 KD = 2.51 ± 0.07), to 231 ± 63 L/kg (2.36 ± 0.1) to 32 ± 5.5 L/kg (1.5 ± 0.08) to 7.5 ± 3.2 L/kg (0.87 ± 0.19) at 0, 0.64, 2, and 16.6 mg C/L added SRNOM (Fig. 1B). Given that >98% of the MeHg was associated with SRNOM for all treatments, it is likely that multiple mechanisms contribute to the observed behavior. One mechanism is the formation of strong aqueous MeHg-DOM complexes that have lower affinity for the MnO2 surface. A second mechanism is competition for limited surface sites between MeHg-SRNOM complexes and SRNOM that is not associated with MeHg. Note that while >98% of the MeHg was associated with SRNOM, <0.21% of the RS groups in SRNOM was associated with MeHg (Supplementary Table S5). These results are similar to those of Muller et al. (2019) in which DOM-sorbent interactions dominated MeHg sorption in PB for four different sorbents.
Isotherm experiments
MeHg sorption isotherms onto MnO2 in the absence and presence of 2 mg C/L SRNOM and in ambient water from EFPC were measured. The creek water had similar pH and DOC concentration to the experiments in PB (Supplementary Table S2), but differs in that it contains other alkali (Na = 0.48 ± 0.13 mM and K = 0.08 ± 0.02 mM) and alkaline earth (Ca = 1.1 ± 0.1 mM and Mg = 0.4 ± 0.04 mM) metals that may affect sorption behavior. Nevertheless, the different water composition and pH of EFPC had very minor effects on predicted MeHg aqueous speciation compared to DOM (>99% MeHgRS; Supplementary Table S5 and Supplementary Fig. S1). MeHg-DOM complexes dominated speciation between pH 7–8 in the SRNOM PB solutions and between pH 5.25 and pH 10 in EFPC water (Supplementary Fig. S1).
MeHg sorption isotherms onto MnO2 showed no indication of sorption saturation across a range of initial MeHg concentrations from 5 to 500 ng/L for each condition tested (Fig. 2 and Supplementary Fig. S2). MeHg sorption decreased ∼10 times with the addition of SRNOM and when experiments were conducted in EFPC water, consistent with the previous results (Fig. 1B). Similarly, this suggests MeHg-DOM and DOM-surface interactions dominate the sorption behavior. The Freundlich isotherm had lower AICc and greater relative likelihood than the linear isotherm for each condition, but in no case was the Freundlich isotherm significantly favored over the linear isotherm (Supplementary Table S6) (Wagenmakers and Farrell, 2004).

MeHg sorption isotherms on MnO2 in the absence and presence of SRNOM at 2 mg C/L and when conducted in filtered EFPC water (2.2 mg C/L) for either 1 day or 3 days. Symbols represent observations and solid lines represent the best fit Freundlich isotherm (Supplementary Table S6 for isotherm parameters and fit summary). EFPC, East Fork Poplar Creek.
Environmental Implications
MnO2 has been proposed as a potential sediment amendment for Hg-contaminated systems. Previous investigators have suggested that Mn oxides are strong Hg sorbents and may limit MeHg by making inorganic Hg unavailable for methylation (Farrell et al., 1998), catalyzing abiotic demethylation (Jackson, 1989), or poising the redox potential at values that are not conducive to methylation (Vlassopoulos et al., 2018). The amorphous MnO2 used in this study did not catalyze demethylation over the 72 h studied. The solid:water partition coefficients (KD) were two orders of magnitude lower than those reported for some engineered sorbents such as SediMite, biochar, and Thiol-SAMMS in phosphate buffer at pH = 7.1–7.5 with I = 0.17 M (Muller et al., 2019). In the absence of SRNOM, MnO2 was a better sorbent than Organoclay-199, but the order was reversed when DOM (either SRNOM or present in natural creek water) was present. Similar to other MeHg and Hg sorbents, increasing concentrations of SRNOM strongly decreased sorption to MnO2. Increasing ionic strength resulted in minor decreases in sorption.
The amorphous form of MnO2 used in this study is not as effective as other common sorbents under environmentally relevant conditions. However, the modest sorption of MeHg to MnO2 could be an added benefit if MnO2 is applied with the purpose of inhibiting methylation by manipulating redox conditions in the contaminated sediment. Future work is needed to determine whether different MnO2 mineral phases have different sorption or demethylation properties. MnO2 is unique relative to many other sorbents in that it is redox active. In this study, we did not address the potential of MnO2 to react with DOM. Future research could investigate MnO2 reaction with DOM and the impact of DOM oxidation on MeHg-DOM complexes and sorption. Finally, it is unclear whether sorbed MeHg is bioavailable to benthic organisms. Further work is necessary in this area before the wide-scale adoption of sorbent amendments for MeHg remediation.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the U.S. Department of Energy, Office of Science, Biological and Environmental Research, Subsurface Biogeochemical Research (SBR) Program, the U.S. Department of Energy's Oak Ridge Office of Environmental Management (OROEM), and URS | CH2M Oak Ridge LLC (UCOR). This research was supported, in part, by an appointment to the Oak Ridge National Laboratory HERE Program, sponsored by the U.S. Department of Energy and administered by the Oak Ridge Institute for Science and Education. The isotopes used in this research were supplied by the United States Department of Energy Office of Science by the Isotope Program in the Office of Nuclear Physics.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.