
Abstract
Currently, a vast majority of bacteria in the natural environment cannot be cultured or isolated under laboratory conditions mainly due to interdependence and interaction of cells. Quorum sensing (QS) is a cell-to-cell communication through which microbial cells regulate their biological activities. In this study, the interfacial sediments from Lake Taihu were induced by three types of signal compounds, including N-butanoyl-L-homoserine lactone (C4-HSL), autoinducer-2 (AI-2), and cyclic adenosine monophosphate (cAMP) under the laboratory conditions, and the variations in microbial diversity and composition after induction were investigated by culture-independent and culture-dependent methods, respectively. High-throughput sequencing analysis revealed that the dominant genera Vogesella and Cloacibacterium in the original sediment were supplanted by Aeromonas and Comamonas genera in response to AI-2 addition. However, the major microbial community in sediments induced by cAMP and C4-HSL was basically the same as that of the original sediment. The screen results indicated cAMP-like of Massilia strains and C4-HSL activity of Bacillus strains. Furthermore, cAMP and AI-2 stimulated Microbacterium strains, whereas C4-HSL and AI-2 promoted Arthrobacter. These findings together confirm the role of QS in mediating microbial community diversity and composition.
Introduction
Quorum sensing (QS) is a widespread form of bacterial cell–cell communication, which depends on microbes production, secretion, detection, and response to certain signal molecules (Ramsey et al., 2009; Wu et al., 2020). QS regulates various bacterial physiological and biochemical functions, such as biofilm formation (Hou et al., 2017), virulence mediation (Stein and Schikora, 2018), chitinase production (Kim et al., 2017), clustering ability (Rajput and Kumar, 2017), and flagellar activity (Ilic-Tomic et al., 2016). Generally, QS system in bacteria can be divided into four categories as follows: (1) LuxI/LuxR system that uses acyl-homoserine lactones (AHLs) as signal molecules; (2) LuxS system with autoinducer-2 (AI-2); (3) CqsA/S system that uses cholerae autoinducer 1 as signal molecules; (4) two-component system that uses modified oligopeptides as signal molecules (Zhao et al., 2015). However, the extent to which QS modulates the aforementioned behavioral responses of bacteria is species specific.
AHLs are the predominant signaling molecules used mainly by Gram-negative bacteria (Kai and Bassler, 2016). AHLs primarily drive intraspecies communication; their accumulation in the intercellular environment ensue the activation of specific promoters that results in the expression of genes, such as the production of antibiotics, luminescence, exoenzymes, surfactants, and exopolysaccharides (Valle et al., 2004). AHLs are composed of fatty acyl chains (4–16 carbons) linked to a lactonized homoserine through an amide bond, and N-butanoyl-L-homoserine lactone (C4-HSL) has a higher activation threshold than other QS molecules used in synthetic biology (Goodson et al., 2017), further contributing to the functional diversity of these compounds in bacterial signaling (Stock et al., 2020).
Gram-positive bacteria generally use AI-2 to regulate gene expression and physiological behavior in either intraspecies or interspecies communication. AI-2-mediated signaling pathway is parallel to and integrated with the pathways mediated by AHLs (Zhao et al., 2018). The precursor of AI-2 is (S)-4,5-dihydroxy-2,3-pentanedione, which undergoes catalysis by S-ribosylhomocysteine lyase (LuxS). It combines with borate to form a highly active AI-2. Chen et al. (2002) reported that borate addition to AI-2 precursor generated active AI-2. Furthermore, AI-2 induced resuscitation of viable but nonculturable (VBNC) Vibrio vulnificus (Ayrapetyan et al., 2014).
Microbial community dynamics is also shaped by the ability of bacteria to respond to a variety of abiotic and biotic factors. This sensing response is mediated by the universal second messenger cyclic adenosine monophosphate (cAMP) (Laganenka et al., 2019). Adenylate cyclases (ACs) convert adenosine triphosphate (ATP) to cAMP. Bruns et al. (2002) reported the role of cAMP in the regulation of many genes, which significantly enhanced the cultivation success of bacterioplankton from the Baltic Sea.
Nowadays, the cultivable rate of environmental bacteria is only about 1% due to the limitations of traditional microbial cultivation methods, whereas the improvement the bacterial cultivation efficiency is meaningful for the development of bacterial germplasm resources. The blocking of information exchange between bacteria is an important factor that limits the bacterial culture efficiency, and QS are called the “language” of bacteria and can regulate the communication between bacteria. Moreover, several reporters had employed the signaling molecules to screen for bacteria that respond to QS. However, these studies only provided or preliminary identification of evidence that bacteria can respond to QS, and the role of QS in mediating microbial diversity and composition remains to be further studied.
Therefore, we aim to explore the influence of three different kinds of exogenous signaling molecules: cAMP, AI-2, and C4-HSL, on the microbial composition and diversity of the interfacial sediment from Lake Taihu under the laboratory conditions rather than natural environments. In this study, we investigated the bacterial community structure using culture-independent high-throughput sequencing and identified the dominant bacteria using a culture-dependent method. This study will help to reveal the role of QS in regulating microbial diversity and communities, and provide a basis for the development and utilization of microbial resources.
Materials and Methods
Sediment sampling
In October 2016, the interfacial sediment sample (top 0–10 cm) was collected in the Zhushan bay (31°27′0.020″ N, 120°48′70″ E) of Lake Taihu (Jiangsu province, China) (Fig. 1). Sediment sample was transported to the laboratory at 4°C and was analyzed immediately. The physicochemical characteristics are presented in Table 1.
Location and description of Lake Taihu.
The Physicochemical Characteristics of Interfacial Sediment from Lake Taihu
IP, inorganic phosphorus; OP, organic phosphorus; TN, total nitrogen; TOC, total organic carbon; TP, total phosphorus.
QS substance add-in test
In this study, the interfacial sediments were induced by three types of signaling molecules, cAMP, AI-2, and C4-HSL, under the laboratory condition that may be unlikely occur in the environment. Boric acid (H3BO3) was used as the AI-2-based signaling substance for its important role in AI-2 synthesis process. The 250 mL sealed flasks, which were filled with 10 g of interfacial sediment and 90 mL of sterilized R2A liquid medium, were placed in the anaerobic tank. R2A liquid medium (g/L): yeast 0.5, starch 0.5, tryptone 0.5, glucose 0.5, casein acids hydrolysate 0.5, MgSO4·7H2O 0.05, C3H3NaO3 0.3, K2HPO4·3H2O 0.3; pH 7.0–7.2 (Reasoner and Geldreich, 1985). Different doses of cAMP (0, 1, 10, 20, and 50 μM), C4-HSL (0, 1, 10, and 20 μM), and H3BO3 (0, 20, and 200 mM) were added to the R2A liquid medium. Flasks were incubated in a shaker table at 28°C for 1 week, while shaking at 150 rpm intermittently to make the bacteria in interfacial sediment sample contact with the signal compounds more fully. The supernatants were collected daily, and the effect of signaling molecules on cell growth was determined by measuring the optical density (OD600) of the culture medium. The experiments were performed in triplicate.
High-throughput sequencing
The interfacial sediment samples induced by 10 μM cAMP, 10 μM C4-HSL, and 200 mM H3BO3, for 7 days were centrifuged at 8,000 rpm, and the precipitate was used for high-throughput sequencing in triplicate. Total genomic DNA was extracted using the E.Z.N.A. Soil DNA isolation kit (OMEGA). The qualities of extracted DNA were examined using 1% agarose gel electrophoresis and quantified by Qubit® ssDNA Assay Kit. Afterward, DNA were amplified by polymerase chain reaction (PCR) using primer set 338F (5′-ACT CCT ACG GGA GGC AGC A-3′) and 806R (5′-GGA CTA CHV GGG TWT CTA AT-3′) for the V3–V4 region of 16S rRNA genes, following the procedures described by Qu et al. (2019). PCR amplicons were assessed using 2% gel electrophoresis, and purified by the SanPrep Column DNA Gel Extraction Kit. A composite sample was mixed approximately equal amounts of purified PCR products from each sample, and then sequenced by Illumina Miseq 2500 platform.
After sequencing, 73,500–75,600 raw sequences were obtained for each sample. Afterward, the barcodes, primers, low-quality sequences, and chimeras were removed using Mothur software, and 60,000 effective sequences were yielded for each sample with an average read length of 444 bp. The raw sequences were deposited into the NCBI short reads archive (SRA) database under accession number of PRJNA612166. The effective sequences were clustered into operational taxonomic units (OTUs) based on a 97% similarity using quantitative insights in microbial ecology. Alpha diversity indices and Good's coverage were calculated and analyzed using Mothur software. Phylogenetic affiliation of each 16S rRNA gene sequence was performed using the Ribosomal Database Project (RDP) classifier. Cluster analysis was performed using the Primer 6 software. Principal component analysis (PCA) was performed using Canoco 4.5 software.
Bacterial isolation and phylogenetic analysis
The interfacial sediment samples induced by 10 μM cAMP, 10 μM C4-HSL, and 200 mM H3BO3 for 7 days, were used for bacterial isolation. The suspensions were 10-fold serially diluted in 0.9% saline solution and plated on the R2A medium at 28°C for 14 days. The morphology and total number of bacteria were observed and counted every day. Bacterial colonies with different morphology were selected and purified.
The DNA of purified bacteria aforementioned was extracted and used as the template for the amplification of 16S rRNA genes, using the universal primers 27F and 1492R (Moreno et al., 2002). PCR reaction mixture contained 0.5 μL of each primer, 12.5 μL of premix Taq, 0.5 μL of template DNA, 11 μL of ddH2O. PCR protocol included initial denaturation at 94°C for 3 min, followed by 30 cycles of 94°C for 45 s, 58°C for 45 s, and 72°C for 2 min, with a final extension at 72°C for 10 min. The amplicon products were visualized on agarose gels (0.8%) and then sent to Sangon Company (Shanghai, China) for sequencing. Bacteria with different sequences in the three parallel samples of the four treatment groups are defined as the typical strains. The homologous sequences were determined in the NCBI Genbank by performing the BLAST. The aligned 16S rRNA gene sequences were constructed by MEGA 7.0 software using the neighbor-joining method, and phylogenetic analyses were conducted with maximum likelihood method.
Analytical methods
Total phosphorus (TP) and total nitrogen (TN) concentrations were analyzed according to the Standard Methods for Water and Wastewater Examination (APHA, 2012). The P fractions in sediment samples were estimated using the Standards Measurements and Testing Program (SMT protocol) as described in Ruban et al. (2001). The pH was measured using a pH meter (HQ30d, HACH, America). Total organic carbon (TOC) concentration was determined using a TOC analyzer (TMM-1, Shimadzu, Japan). All measurements were performed in triplicate unless otherwise indicated. All statistical analysis was performed using analysis of variance (ANOVA) (Statistical Product and Service Solutions, version 18.0) with a significance level of p < 0.05.
Results and Discussion
Optimal dosage of QS substance
The fluctuations in cell-population density under different doses of cAMP, C4-HSL, and H3BO3 are shown in Supplementary Fig. S1. The cell-population density fluctuated greatly with the increase of cultivation time when the cAMP was added into the interfacial sediment. Compared with the control group, its population density reached its maximum value on the fourth day with the 10 μM of cAMP addition, whereas it declined with 20–50 μM cAMP addition. Meanwhile, 10 μM C4-HSL addition increased the cell population density, especially from days 3 to 7. The cell population density is basically same with the control group when the dosage of H3BO3 addition was 2 mM, whereas it increased rapidly with the 200 mM H3BO3 addition. These results indicated that 10 μM cAMP, 10 μM C4-HSL, and 200 mM H3BO3 are contributed to the increase of microbial density in the sediment.
Microbial community analysis by culture-independent method
Richness and diversity of bacterial community
The interfacial sediment samples, which were induced by 10 μM cAMP, 10 μM C4-HSL, and 200 mM H3BO3, were used for microbial community analysis by high-throughput sequencing. The coverage values of all four samples ranged from 99.71% to 99.85% (Table 2), which suggest that the sequence libraries constructed in this study adequately accounted for the bacterial community. The OTU numbers retained from the four samples varied from 484 to 540, of which 234 OTUs were shared by all the 12 samples (Supplementary Fig. S2). This finding indicates that the microbial community structure in the interfacial sediment shifted after induction by cAMP, C4-HSL and AI-2. Chao1 and ACE indices are positively related to microbial richness, and Shannon and Simpson indices are commonly used to characterize species diversity in the bacterial community. The bacterial richness in sediments induced by the three signaling molecules decreased, whereas the bacterial diversity in sediment induced by C4-HSL exhibited the highest bacterial richness compared with the control.
Biodiversity Indices Of Four Samples (K: Origin Sediment; A, C, H: Sediment Treated by cAMP, C4-HSL, AI-2)
AI-2, autoinducer-2; C4-HSL, N-butanoyl-L-homoserine lactone; cAMP, cyclic adenosine monophosphate.
Taxonomic complexity of bacterial community
Bacterial structure succession in the interfacial sediment samples are shown in Fig. 2. At the phylum level, Proteobacteria and Bacteroidetes were numerically the dominant phyla (91% of the total sequences) in all the 12 samples, which are in accordance with the previous reports on eutrophic lake bacterial community (Chen et al., 2017). Although the main phyla were the same across the interfacial sediment samples induced by cAMP (A), C4-HSL (C), and AI-2 (H), there were differences in their relative abundances. Compared with the control group (K), the relative abundance of Proteobacteria increased with cAMP, C4-HSL, and AI-2 treatments, whereas the relative abundance of Bacteroidetes decreased. As the most abundant phylum, Proteobacteria increased by 3.64% (A), 3.52% (C), and 5.1% (H), which indicated that there may exist widespread QS as the majority of micro-organisms with AHL-based QS related genes belonging to Proteobacteria (Case et al., 2008). Whereas Bacteroidetes decreased by 3.14% (A), 3.87% (C), and 3.77% (H) compared with the control. Moreover, Chlorobi gradually decreased from 1.88% (K) to 0.24% (H), whereas Firmicutes increased from 2.17% (K) to 4% (H). This finding is in accordance with the reports by Pereira et al. (2009) who demonstrated the presence of AI-2-mediated QS in various members of the phylum Firmicutes.
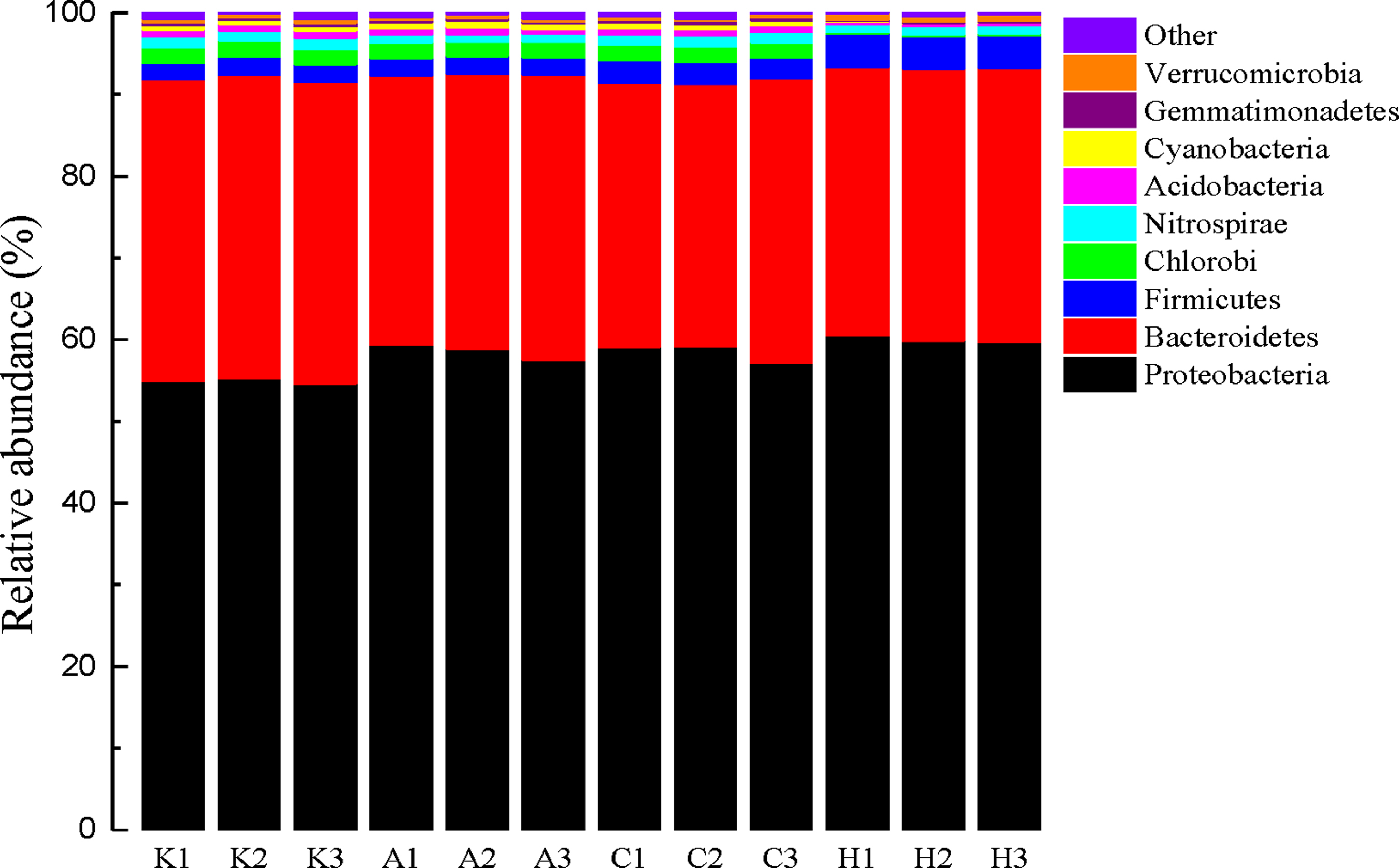
Taxonomic classification of bacterial communities at phylum level. The relative abundance <0.3% in all the four samples was defined as “others” (K: origin sediment; A, C, H: sediment treated by cAMP, C4-HSL, AI-2). AI-2, autoinducer-2; C4-HSL, N-butanoyl-L-homoserine lactone; cAMP, cyclic adenosine monophosphate.
At the genus level, the relative abundance of bacteria differed among the sediment samples induced by cAMP, C4-HSL, and AI-2 (Fig. 3a). Vogesella was the most abundant in control (K), and its abundance in AHL-treated and cAMP-treated groups was consistent with that of control; however, it was almost undetectable in AI-2-treated sediment. Flavobacterium was the second most abundant genus in K, its abundance dropped slightly (0.6–0.7%) in A and C, whereas it increased obviously in H (4.6%); this phenomenon revealed that the effect of AI-2 on Flavobacterium might be stronger than exogenous AHL and cAMP. In contrast, Aeromonas, Comamonas, and Pseudomonas enriched greatly in H, and their relative abundance increased 14.2%, 12.4%, and 3.9%, respectively, compared with the control. These genera turned into abundant indicated that the exogenous QS was favorable to promote the communication between these bacteria. Furthermore, Cloacibacterium, Chitinophaga, and Bdellovibrio were less abundant in A, C, and H than that in K. The mechanisms of QS were very complex, QS molecules produced and released by certain bacteria could have an influence on bacteria from the same group or other groups, whereas some bacteria may use more than one type of QS substance to communicate with each other (Ouyang et al., 2020).

Bacterial composition at genus level
The composition of the bacterial genera in H was the most distinct in comparison with K, A, and C. Six genera showed significant differences among the four treatment groups (Fig. 3b). There was a significantly lower abundance of Vogesella (p < 0.01), Steroidobacteraceae (p < 0.01), and Chitinophagaceae (p < 0.01) in H. Meanwhile, the relative abundance of Aeromonas (p < 0.01), Bacillus (p < 0.05), and Hydrogenophaga (p < 0.05) was significantly higher in H.
Based on pyrosequencing data, PCA with weighted uniFrac distance and clustering analysis were performed in Fig. 4. The PCA score plot indicated that K, A, and C were closely related and were grouped to the right of the graph along PC2, which represented 23.43% of the total variation. Whereas H remained separated from K, A, and C groups along PC1, which accounted for 68.91% of the total variation. Besides, the cluster analysis also confirmed that the bacterial community in H was significantly different from that of K, A, and C.
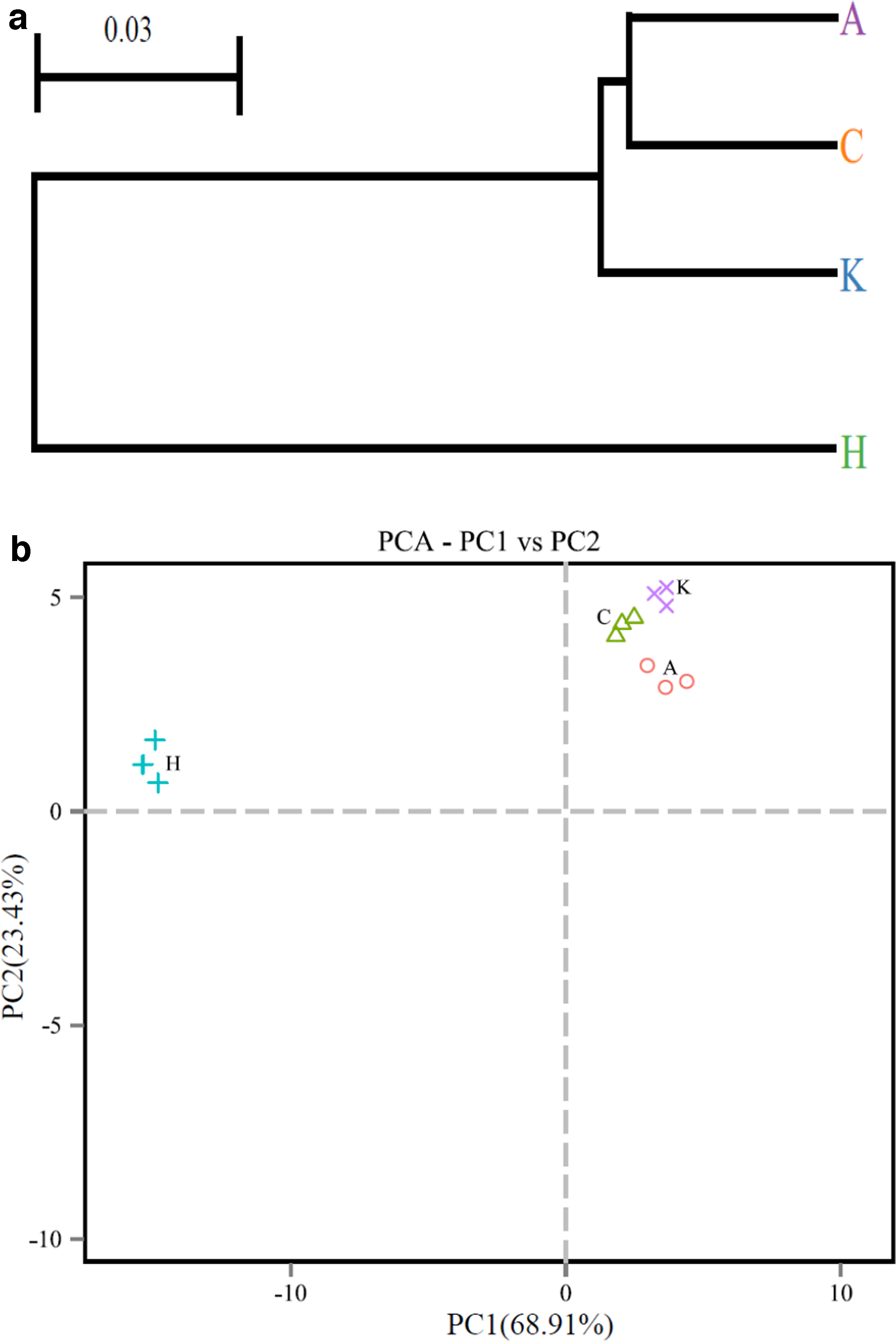
Beta diversity: cluster analysis
Microbial composition analysis by culture-dependent method
After selection and enrichment with cAMP, C4-HSL, and AI-2, a total of 33 active bacterial strains were obtained from interfacial sediments (Supplementary Table S1). The maximum cultivation bacterial count was attained in the sediment supplemented with cAMP, whereas minimum was with C4-HSL. Among them, C4-HSL and AI-2 performed well with the highest number of bacterial strains. 16S rRNA-based phylogenetic trees were constructed for the isolates and their close relatives according to GenBank. As illustrated in Fig. 5, these isolates belonged to Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. At the genus level (Fig. 6), control group was dominated by Bacillus, Pseudomonas, and Flavobacterium genera. Bacillus and Pseudomonas had an absolute quantitative majority in C4-HSL and AI-2 groups, respectively, whereas the abundance of Flavobacterium decreased when induced by cAMP, C4-HSL, and AI-2. Furthermore, sediment treated with cAMP showed preference for rare bacterial strains in the isolation process; Massilia and Microbacterium were dominant. Enterobacter, Bosea minatitlanensis, Trichococcus, and Lysinibacillus were identified only in H, indicating that AI-2 might play an important role in recovery of those genera.
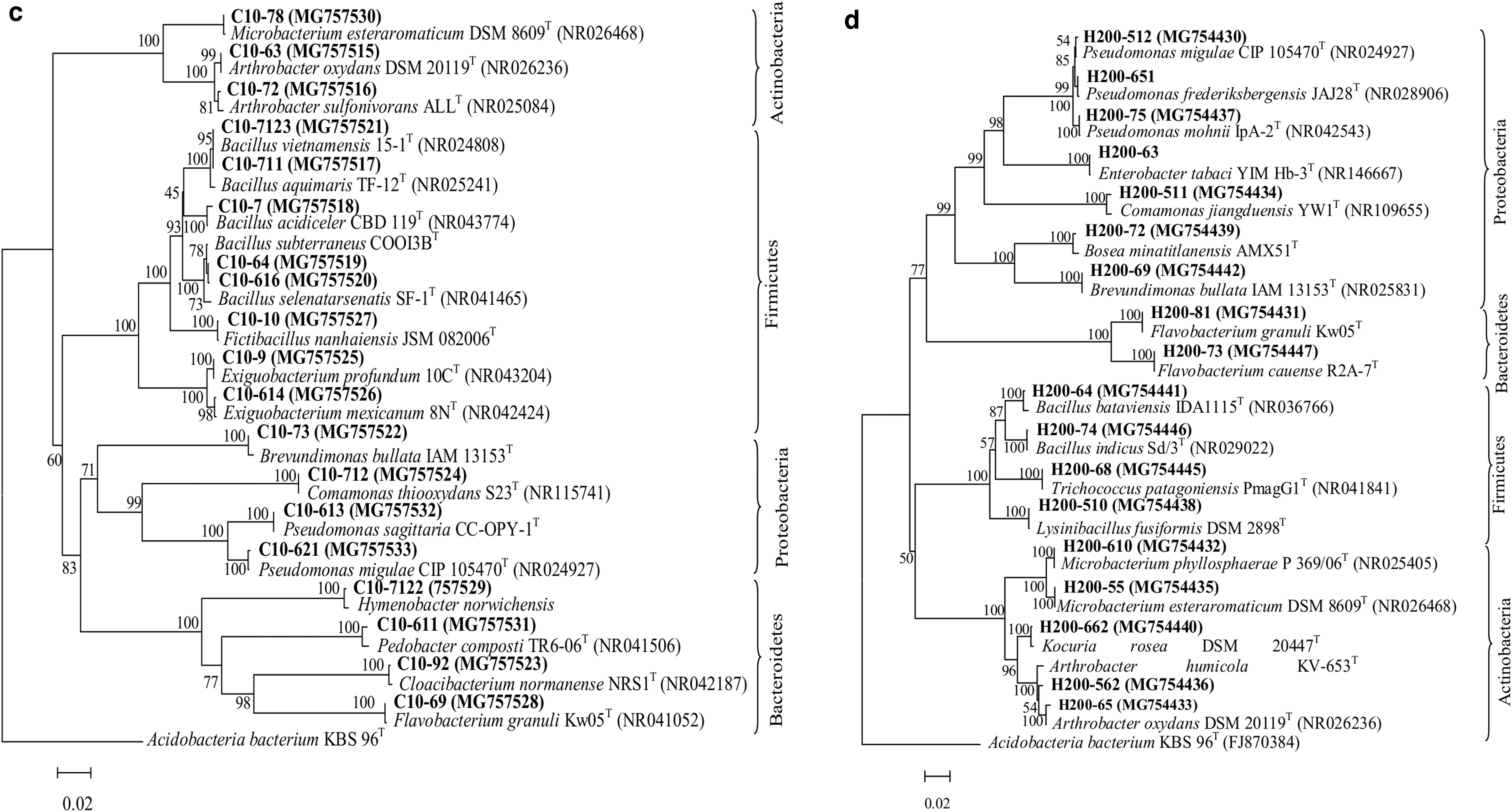
Phylogenetic relationships of bacterial communities isolated from the control treatment
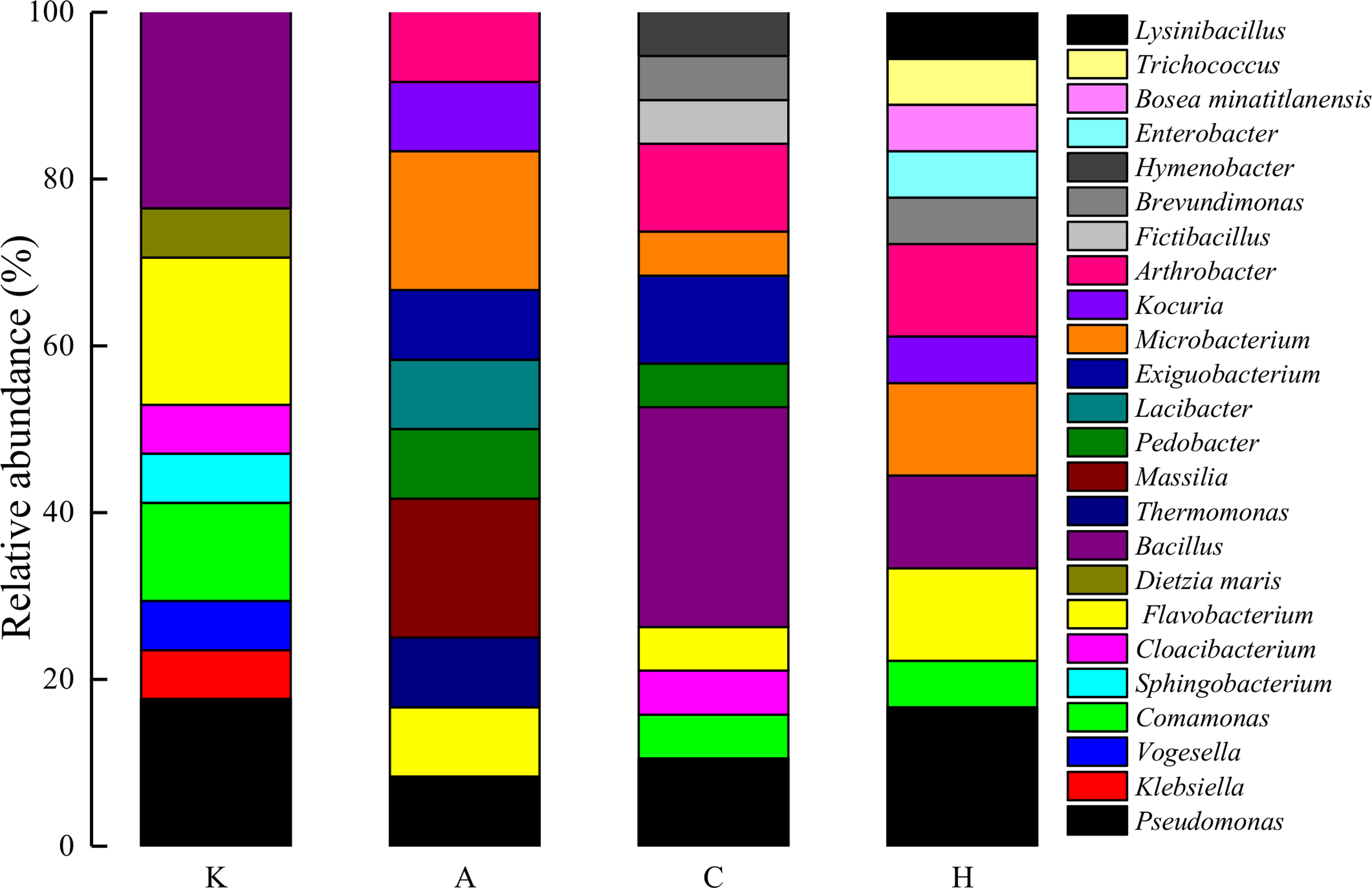
Taxonomic classification of isolated bacteria at genus level (Refer to Fig. 2 for sample abbreviations).
Comparison between the culture-independent and culture-dependent methods
The addition of cAMP, C4-HSL, and AI-2 in lake sediment influenced bacterial richness and diversity; however, no obvious difference was observed in the dominant bacterial phylum between the treatment groups and the control. Furthermore, microbial composition analysis by both culture-independent and culture-dependent methods revealed the same results (Table 3); Proteobacteria and Bacteroidetes were the dominant phyla in the four sediment samples.
Comparison of Microbial Communities by Culture-Dependent and Culture-Independent Methods

Identification of Pseudomonas and Flavobacterium in the four samples by both culture-dependent and culture-independent methods indicates that the dominant bacteria in the sediment could be cultured under laboratory conditions. The relative abundance of Pseudomonas analyzed by culture-dependent and culture-independent method both decreased in the sediment induced by cAMP and C4-HSL, whereas the abundance analyzed by culture-independent method was similar to that in control and that analyzed by culture-independent method in H. Studies have reported that elevated levels of cAMP and C4-HSL inhibited biofilm formation of Pseudomonas aeruginosa through Vfr and rhl (Almblad et al., 2019; Zhao et al., 2019). Furthermore, genus of Pseudomonas has the ability of AI-2 producing and sensing, which may be conducive to its enrichment in AI-2-treatment group (Høyland-Kroghsbo et al., 2016).
High-throughput sequencing revealed Vogesella and Cloacibacterium as the dominant genera in K, A, and C; however, only one Vogesella strain was isolated from K, and two Cloacibacterium strains were isolated from K and C. This phenomenon may be partly due to the fact that many bacteria in the natural environment are VBNC (Su et al., 2013). In such a state, bacteria fail to grow on conventional media under laboratory conditions due to low metabolic activity.
Aeromonas, Bdellovibrio, and Chitinophaga were detected only by culture-independent method, and the abundance of Aeromonas in the sediments treated by cAMP, C4-HSL, or AI-2 was more than that in control. Aeromonas possesses an AHL-dependent QS system based on ahyRI locus, and previously, Nagar et al. (2015) had demonstrated that C4-HSL was the major AHL molecule of Aeromonas hydrophila. Hence, the addition of C4-HSL in the sediment promoted Aeromonas enrichment. The abundance of Aeromonas increased significantly (p < 0.01) in response to AI-2 treatment. This finding confirms the reports by Martins et al. (2018) who demonstrated that AI-2 strongly induced the QS in A. hydrophila.
Arthrobacter and Microbacterium were recovered only by the culture-dependent approach from cAMP, C4-HSL, and AI-2 groups. Microbacterium and Arthrobacter are the AHL-producing bacteria (Waheed et al., 2016; Yavuztürk and Koyuncu, 2017), and they have the ability to produce cAMP and possess AI-2 activity (Cao et al., 2012; Gori et al., 2011). In this study, cAMP, C4-HSL, and AI-2 are employed as signals to rouse the dormant population and, therefore, addition of these signal compounds yielded more number of cultivable members of Arthrobacter and Microbacterium.
Culture-dependent and culture-independent methods identified Comamonas and Bacillus in K, C, and H, whereas only culture-independent method identified these in A group. Partial AHL QS-related genes were harbored by Comamonas (Feng et al., 2019), then involved in AHLs metabolism. Bacillus is also one of the AHL-producing bacteria. Cao et al. (2012) isolated AHL-degrading bacteria, mostly Bacillus, from the natural environment. This could explain the predominance of Bacillus in C detected by culture-dependent and culture-independent methods.
Conclusions
In this article, the influence of C4-HSL, AI-2, and cAMP supplementation on bacterial composition and activity of lake sediment was systematically studied by culture-independent and culture-dependent methods. High-throughput sequence analysis revealed no significant difference in the dominant bacteria among the cAMP and C4-HSL treatment groups and the control group; however, AI-2 addition positively regulated Comamonas and Aeromonas. The cultured analysis indicated that Massilia and Bacillus strains displayed cAMP-like and C4-HSL activity, respectively. cAMP and AI-2 addition positively regulated Microbacterium strains, and AI-2 promoted the culturability of Arthrobacter. To conclude, this study highlights that signal compounds are essential for bacterial culture.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.