
Abstract
Control of toxic or carcinogenic disinfection byproduct (DBP) formation in drinking water is critical in an effort to improve drinking water safety and safeguard public health, due to the elevation of dissolved organic matter in source water. In this study, the oxidation by ferrous iron (Fe2+) and hydrogen peroxide (H2O2) reactions was investigated to assess its efficacy for reducing the DBP forming potentials of four trihalomethanes (THMs) and five haloacetic acids (HAAs) that are currently regulated by the United States Environmental Protection Agency. Results indicated that the oxidation efficacy was dependent on pH, concentrations of Fe2+ or H2O2 added, and initial dissolved organic carbon (DOC) (Resorcinol used as a model compound) level in simulated source waters. Under pH 5.0 and initial 2 mg C/L conditions, the treatment using 0.25 mM of Fe2+ and H2O2 was able to achieve the reductions of 94% THMs and 77% HAAs forming potentials after 90 min reaction. Furthermore, the treatment would also lead to 30% and 36% decreases in DOC and chemical oxygen demand, respectively. More importantly, the oxidation reactions showed the similar reduction efficiency for the DBP forming potentials in the presence of Escherichia coli and resulted in the effective E. coli disinfection in drinking water. pH was identified as one of the most important parameters affecting the efficacy for DBP control. This research demonstrated that the oxidative treatment by the Fe2+-H2O2 reactions would effectively mitigate DBP formation through oxidative removal of DOC and achieve drinking water disinfection simultaneously as well, which could be potentially applied, as a cost-effective and environmental-safe drinking water treatment technology, to small water systems in rural communities with elevated Fe2+ and slightly acidic source water.
Introduction
The World Health Organization (WHO) reported that there were estimated 2.1 billion people lacking safe drinking water worldwide, especially the people living in rural areas (WHO, 2017). The people living in United States generally have safe drinking water (Rose, 2021). However, according to the latest report by the Centers for Disease Control and Prevention, there were about 42 drinking water-associated disease outbreaks during the 2013–2014 period, responsible for at least 1066 cases of illness, 124 hospitalizations, and 13 deaths (Benedict et al., 2017).
Disinfection through chlorination is the most common practice in drinking water treatment to control the waterborne diseases caused by toxic pathogens. Paradoxically, disinfectants such as chlorine not only deactivate pathogens but also react with dissolved organic matter (DOM) in source water, leading to the formation of numerous disinfection byproducts (DBPs). Many of the DBP compounds have been confirmed as toxic and potentially carcinogenic (Bond et al., 2012). The United States Environmental Protection Agency (USEPA) started to regulate several groups of DBPs, including trihalomethanes (THMs) and haloacetic acids (HAAs), in drinking water since 1970s and has tightened the regulations to lower the health risks of DBPs several times afterward.
DOM, as a precursor for the DBP formation, is normally in a range of 0.1 to a few tens mg/L in surface waters and was reported as constantly increasing due to agriculture operations and human activities (USEPA, 2018). Increased DOM in source water and consequent elevation of DBP in drinking water have been a challenge to small water systems in compliance with the USEPA DBP regulations, because of limited financial resource and technical difficulty. In an effort to improve drinking water safety and safeguard public health, as well as assist drinking water systems in compliance with the DBP regulations, especially small water systems in rural communities, developing more efficient and cost-effective technologies for drinking water disinfection and DBP control is critically needed and has been a research focus over a few decades.
In general, common approaches for the DBP control included: DOM removal as DBP precursors before disinfection and selection of nonchlorine disinfectants. Activated carbon and membrane filtration were the most common approaches for DOM removal, whereas advanced chemical oxidation, including UV-enhanced disinfection, ozonation, peroxyacetic acid or peracetic acid oxidation, and Fenton or Fenton-like reactions are also being studied (Wang et al., 2015; Yang et al., 2018; Water Research Foundation, 2019). There have been considerable interests in applying conventional Fenton or Fenton-like reactions, oxidative reactions of H2O2 decomposition catalyzed by a variety of iron species, for the treatment of water contaminants (Gogate and Pandit, 2004). The hydroxyl radicals (·OH), a strong oxidative species generated by Fenton or Fenton-like reactions at room temperature and ambient atmospheric pressure, are strongly oxidative species and have a higher oxidative capability (2.73 mV redox potential) than HClO generated by chlorination (1.49 mV redox potential) (Buxton et al., 1988). Hydroxyl radicals were reportedly able to react with a variety of environmental refractory contaminants at near diffusion-control rate (Deng and Englehardt, 2006; Wu et al., 2010). In conventional Fenton reaction, ferrous iron was commonly used (Deng and Englehardt, 2006), while other iron forms such as iron oxides or complexed Fe3+ were used in Fenton-like reaction (Lin and Gurol, 1998). Fenton and Fenton-like reactions and the applications have been reviewed in detail by several review and research articles (Pereira et al., 2012; Salgado et al., 2017; Thomas et al., 2021). The majority of Fenton reaction studies were focused on the remediation of environmental contaminants in wastewater or soil, but the application in drinking water DBP control was little reported.
Murray and Parsons (2004) applied Fenton and photo Fenton reactions for the removal of dissolved organic carbon (DOC) in reservoir water and found DOC removal by 90% and a substantial reduction of THM formation potential at pH 4–6. Moncayo-Lasso et al. (2012) reported that photo-Fenton at near neutral pH could be used as a pretreatment of river water to reduce its THM formation. Gosselin et al. (2013) indicated that Fenton and Fenton-like reactions were able to disinfect biofilms under an emergency contamination scenario. Although three studies mentioned showed the potential application of Fenton or Fenton-like reactions in drinking water treatment, there are still several questions needed to be addressed as to its practical applications. For example, what is the reaction mechanism for DBP control by Fenton and Fenton-like reactions? whether the reduction of THM formation is achieved by the DOC mineralization or by the oxidation of DOM reactive functional groups? do Fenton or Fenton-like reactions yield HAAs? and can hydroxyl radicals be efficiently generated under drinking water treatment relevant conditions?
Iron species are naturally present in the range of mg/L in most groundwater or surface water. Ferrous iron at 15 mg/L was measured in the source groundwater in a small drinking water system near Kansas City, MO (Hua et al., 2016), although most river waters contained much less iron. As regulated by the USEPA National Secondary Drinking Water Regulations, total Fe content in finished water from drinking water treatment plant must be below 0.3 mg/L. Most drinking water systems commonly applied compressed air or other oxidants to reduce total Fe by transforming ferrous iron to less soluble ferric oxy(hydroxides) at the beginning of water treatments (Hua et al., 2016). Therefore, if there is a few mg/L level of ferrous iron already present in source water, the treatment by Fenton and Fenton-like reactions could use such ferrous ions in source water and save the operational costs associated with the aeration practices. In addition, the solid ferric oxy(hydroxide) formed could also serve as a coagulant for water treatment and be easily removed at the end of the operation train. Note that ferrous ion at mg/L level was at stoichiometric amounts for the reduction of DBP forming potentials in our experiment design.
In this study, it was hypothesized that the hydroxyl radicals can be generated by Fenton and Fenton-like reactions at circumvent neutral pH range and effectively reduce both THM and HAA formation through the removal of DOC or DBP precursors. The goal of this study was to investigate the efficacy of Fenton or Fenton-like reactions for DOC removal, DBP control, as well as bacteria disinfection, in drinking water at circumvent neutral pH range. Specific objectives were to (1) evaluate the removal efficiency of DOC by Fenton or Fenton-like reactions under drinking water treatment relevant conditions; (2) determine whether the oxidative reactions could significantly reduce THM and HAA formation potentials; (3) examine whether the oxidation could also effectively inactivate Escherichia coli as a disinfection indicator; and (4) to explore the possible treatment mechanisms.
Experimental
Experimental procedures
Batch experiments were conducted to investigate the effects of Fe2+, Fe3+, H2O2, and initial pH on the removals of DOC and chemical oxygen demand (COD) and the formation potentials of selected THMs and HAAs [THM forming potential (THMFP) and HAA forming potential (HAAFP)]. COD was commonly used in quantifying the amount of oxidizable pollutants in water. In this research, DOC was used to characterize carbon that could be mineralized. Many studies indicated that there was no direct correlation between DOC and DBP formation potential (an oxidation process). Thus, COD was adapted in this study to quantify the amount of oxidizable organic moiety or characterize carbon that could be oxidized after the treatment, as a surrogate parameter more closely related to DBP formation potential than DOC. The experiments were performed in 500-mL glass beakers stirred by Floc-Tester (ET 740, Orbeco-Hellige) in dark at 25°C ± 3°C. Simulated DOC-containing source waters were prepared by transferring the desired amounts of resorcinol (C6H6O2, M.W.110) stock solution to the glass beakers and diluted with Milli-Q water (Millipore system, 18.2 MΩ·cm). Resorcinol, one of three isomeric benzenediols (1,3-isomer or meta-isomer), was used as a model DOC compound to mimic DOM in source water. Hydroxybenzone is one of the common functional groups in natural organic matter responsible for the formation of DBPs (Bond et al., 2012). In this study, resorcinol was selected as a surrogate for organic matter for two reasons: (1) resorcinol had been well characterized for THMFP and HAAFP, and (2) initial DOC/COD and THMFP/HAAFP could be easily controlled, which would minimize experimental variations, reduce the complexity of real natural organic matter, and thus make data comparison and interpretation more accurately. The results of this study may not be completely applicable to drinking source waters containing diverse natural organic precursors.
The desired amount of Fe2+ stock solution was then added in three equal stepwise to mitigate pH variation, and the solution pH was adjusted with 0.1 M HCl or 0.1 M NaOH if needed. Fenton or Fenton-like reaction was initiated by adding H2O2 stock solution. After completion of the reaction, 1.0 mL of 2.0 g/L Ca(OH)2 solution and 0.1 M NaOH solution were added to neutralize pH to ∼7.0, and the solution was allowed to sit for 30 min. A measure of 30.0 mL of the supernatant was subsequently sampled, filtered through a 0.45 μm syringe filter (polytetrafluoroethylene, Fisher brand), and adjusted pH to ∼9.0 with 0.1 M NaOH to stop the generation of hydroxyl radical (Muruganandham and Swaminathan, 2004). The solution was stored in refrigerator for 12 h to quench the residue H2O2 before analysis (Su et al., 2011). Each set of the experiments was repeated with a triplication.
The batch experiments were repeated with the presence of E. coli, following the similar procedures above. A few modifications were described as follows: the desired amount of E. coli stock solution was added to the resorcinol stock solution and stirred for 30 min in dark for bacteria acclimation to new conditions. After completion of the reaction, the residual H2O2 was immediately quenched by adding Na2SO3. Then, 1.0 mL solution was taken to a 2.0 mL tube, and bacteria were scattered using a vortex mixer (Touch Mini Vortexer 945410; Fisher Scientific).
Hydrogen peroxide (H2O2, 30%, w/w), starch solution (1%, w/w), sodium hypochlorite (HClO, 10–15%, w/w), boric acid (H3BO3), sodium sulfite (Na2SO3) and resorcinol (Benzene-1,3-diol, C6H6O2, M.W.110) were purchased from Sigma-Aldrich (St. Louis, MO). Ferric chloride hexahydrate (FeCl3·6H2O), ferrous sulfate (FeSO4), hydrochloric acid (HCl, 37.5%, w/w), ferrous ammonium sulfate hexahydrate [Fe(NH4)2(SO4)2·6H2O], 1,10-phenanthroline monohydrate (C12H8N2·H2O), potassium iodate (KIO3), sodium hydroxide (NaOH), ammonium molybdate tetrahydrate [(NH4)6Mo7O24·4H2O], hydroxylamine hydrochloride (HONH2·HCl), calcium hydroxide [Ca(OH)2], potassium iodide (KI), potassium chloride (KCl), sodium chloride (NaCl), tryptone, yeast, and agar were from Fisher Scientific (Fair Lawn) and DPD free chlorine reagent from Hach Company (Loveland). Sodium hypochlorite and ammonium molybdate tetrahydrate were reagent grade and all other reagents analytical grade. All chemicals were used as received without further purification and glassware for bacterium tests autoclaved at 121°C for 15 min before use.
Analytical methods
The concentrations of ferrous iron (Fe2+) and ferric iron (Fe3+) were determined by 1, 10-phenanthroline colorimetric method (Eaton et al., 1998), with 0.1 mg/L detection limit. The concentration of H2O2 was analyzed using iodide method (Graf and Penniston, 1980), with a detection limit of 0.3 mg/L. COD was measured with a COD Measurement Kit (Hach, DRB 200 digestion chamber, MD 600, Lovibond). DOC was determined by a total organic carbon (TOC) analyzer (TOC-VCSH; Shimadzu, Columbia, MD), with four-point calibrations and 0.1 mg C/L detection limit. Before injection for DOC analysis, the sample was filtered with syringe filter (0.45 μm, polytetrafluoroethylene, Fisher brand) to eliminate bacteria and small particles.
Determination of THMFP and HAAFP
THMFP and HAAFP were determined following the EPA standard method 510.1 (Clements et al., 1987). Briefly, 61.8 g boric acid (1.0 M) and 4.4 g NaOH (0.11 M) were added in 1 L Milli-Q water to prepare a buffer solution (pH 8.3 ± 0.1). A free Cl2 stock solution was prepared by dissolving sodium hypochlorite in the buffer (∼3500 mg/L). The stock solution was prepared weekly to ensure pH at 8.3 ± 0.1 and free Cl2 concentration in the 3000–4000 mg/L range. Before chlorination, samples were diluted with Milli-Q water to bring DOC content to below 3.0 mg/L and pH adjusted to 8.3–8.7 with NaOH (0.1 M) or HCl (0.1 M). The aliquots of the water sample were subsequently transferred to 40-mL amber vials and dosed with the free Cl2 stock solution (0.1–0.3 mL) to ensure free Cl2 in a 2–4 mg/L range after chlorination (monitored by the Hach DPD colorimetric method). To prevent a loss of volatile THMs and HAAs, the sample vials were capped immediately without any headspace after Cl2 dosing, wrapped with aluminum foil to eliminate light exposure, and stored in an incubator for 7 days at 25°C. After the incubation, the samples were quenched by adding 50 μL 0.2 M sodium sulfite to remove any free Cl2 residual.
THMs formed in the sample were analyzed by a rapid and sensitive solid phase microextraction—gas chromatography with electron capture detector method developed by Shi and Adams (2012) with slight modification. For SPME sampling, a 10 mL aliquot of sample was transferred to a 20 mL glass GC headspace vial and then sealed with a Teflon-faced septum. Headspace SPME extraction was performed for 3 min in the vial headspace with a Supelco polydimethylsiloxane fiber with 100-mm coating thickness (Sigma-Aldrich). Desorption was carried out in a GC injection port at 220°C for 3 min in a splitless mode. GC (Agilent 7890A gas chromatography) equipped with an electron capture detector (Agilent Technologies, Inc., Santa Clara, CA) and VOCOL GC column (10 m × 0.2 mm with 1.2 μm film; Supelco, Bellefonte, PA) were used for THM separation. Column oven temperature program was selected as follows: initial temperature was set at 40°C, held for 2 min, then ramped at 20°C min−1 to 150°C, and held for 2 min. The carrier gas (helium) flow rate was 0.7 mL/min. The ECD detector temperature was set at 250°C. The detection limits were 0.20 μg/L for CHCl3 and CHBr3 and 0.05 μg/L for CHBrCl2 and CHBr2Cl.
HAAs were analyzed by a 4000Q Trap mass spectrometer (AB Sciex, Foster City, CA) hyphenated with an ultrafast performance liquid chromatography system, including two pumps (LC-20AD XR), an autosampler (SIL-20AC XR), and an online degasser (DGU-30A3) (Shimadzu), following a high performance ion chromatography–tandem mass spectrometry (HPIC-MS/MS) method (Xue et al., 2016). An aliquot of quenched water sample was filtered using 0.22 μm nylon membrane syringe filter and injected directly for HPIC-MS analysis without any further sample preparation. The IC column was a Dionex IonPac AS21 (2 × 250 mm) with an AG 21 guard column (2 × 50 mm). Mobile phase A was water and B was 200 mM methylamine in water. The elusion flow rate was 0.3 mL/min with the same gradient elution program as described in the method. The mass spectrometer conditions were also same as described in the method (Xue et al., 2016). The quantification detection limits for the HAAs were 0.1 μg/L for monochloroacetic acid, 0.02 μg/L for dichloroacetic acid and monobromoacetic acid, 0.05 μg/L for dibromoacetic acid, and 2 μg/L for trichloroacetic acid.
Quantification of E. coli
Nonpathogenic E. coli strain K12 (MG1655) was selected, as a bacteria indicator of enteric pathogens, for the disinfection experiments. The preparation and plating of E. coli K12 followed the established procedures (Spuhler et al., 2010). Specifically, Luria Bertani (LB) broth was prepared by adding 10.0 g tryptone, 10.0 g NaCl, and 5.0 g yeast extract in 1.0 L Milli-Q water and a saline solution by dissolving 8.0 g NaCl and 0.8 g KCl in 1.0 L Milli-Q water. All solutions were sterilized by autoclaving at 121°C for 15 min. The precultivation and grown culture procedures were performed aerobically. Colony forming units (CFU) were determined by pouring-plating on LB agar (LB broth with 1.5% agar) plates. Preculture of E. coli was conducted by streaking out a loop full from the strain sample onto LB plates, followed by incubating the plates at 37°C for 24 h (Incubator 203FS; Fisher Scientific). E. coli K12 was inoculated from preculture into 5 mL LB and incubated at 37°C for 8 h (Shaker Incubator MAXQ 4450). The solution was then diluted (1% v/v) in 25 mL prewarmed LB broth and incubated at 37°C for 15 h. Cell bacterial pellets were harvested by centrifugation for 15 min at 5000 RCF (Centrifuge ST 16R; Thermo Fisher). The final bacterial pellets were washed with the saline solution twice and resuspended to initial volume. The solutions were diluted in 10% stepwise and plated on LB agar plates for 24 h incubation at 37°C until measurement.
Results
Effect of initial Fe2+ concentration
The effects of increasing initial [Fe2+]0 on the reductions (c/c0) of DOC, COD, THMFP, and HAAFP are presented in Fig. 1. The range of [Fe2+]0 was from 0.0 to 0.5 mM, and the reaction time was 90 min at 25 ± 3°C. The initial reaction conditions were set as DOC = 2.0 mg/L, [H2O2]0 = 0.25 mM, and pH = 5.0, with initial measurements (c0) of THMFP = 3212 μg/L, HAAFP = 84 μg/L, and COD = 15.6 mg/L. As data shown in Fig. 1, all four measurements had a similar decreasing trend, but with different reduction percentages. When [Fe2+]0 was raised from 0 to 0.12 mM, there was no substantial decrease measured in DOC, COD, or HAAFP; nevertheless, there was nearly 40% reduction measured for THMFP. In [Fe2+]0 range from 0.12 to 0.25 mM, all four measurements showed a significant reduction. The reductions of THMFP and HAAFP were 94% and 77%, respectively, while 30% and 36% in DOC and COD. When [Fe2+]0 reached above 0.25 mM, all reductions were relatively steady and not significant. Data indicated that the most effective reductions by [Fe2+]0 would be at 0.25 mM, and higher reductions for THMFP and HAAFP were achieved.
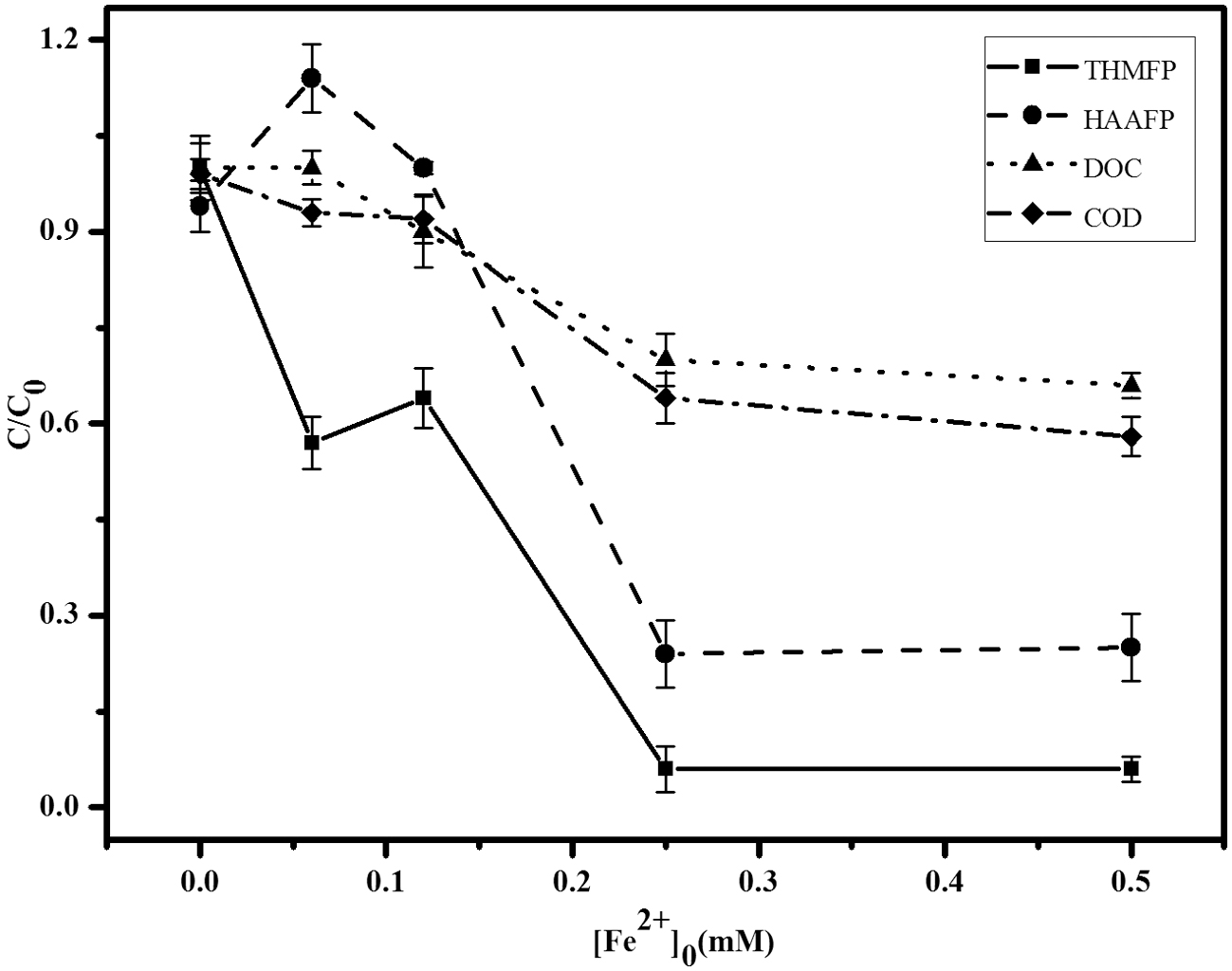
Effect of [Fe2+]0 on measurements of THMFP, HAAFP, DOC, and COD after 90 min reaction time (Error bar indicates standard variation, the initial conditions: 2.0 mg/L DOC (resorcinol), 0.25 mM [H2O2]0, and pH 5.0, the initial measurements (c0) of THMFP 3212 μg/L, HAAFP 84 μg/L, and COD 15.6 mg/L). COD, chemical oxygen demand; DOC, dissolved organic carbon; HAAFP, HAA forming potential; THMPF, THM forming potential.
Effect of initial H2O2 concentration
With similar initial reaction conditions, but constant 0.25 mM [Fe2+]0 and 0.0 to 0.5 mM [H2O2]0 range, increasing [H2O2]0 resulted in a different decline trend of THMFP and HAAFP from that of DOC and COD (Fig. 2). As [H2O2]0 increased from 0.0 to 0.06 mM, there were 22% and 20% reductions for THMFP and COD measured, but neither HAAFP nor DOC reduction was substantial. When [H2O2]0 reached to 0.25 mM, all reductions were shown significant, with THMFP decrease by 94%, HAAFP 77%, DOC 27%, and COD 32%. In contrast to Fig. 1, with [H2O2]0 further increased to 0.5 mM, DOC and COD declines were leveled off, and THMFP and HAAFP started to rebound, dropping the reductions to 72% and 22%, respectively. Similar to data presented in Fig. 1, the treatment by [Fe2+]0 and [H2O2]0 at 0.25 mM would achieve the best reductions for all parameters measured.
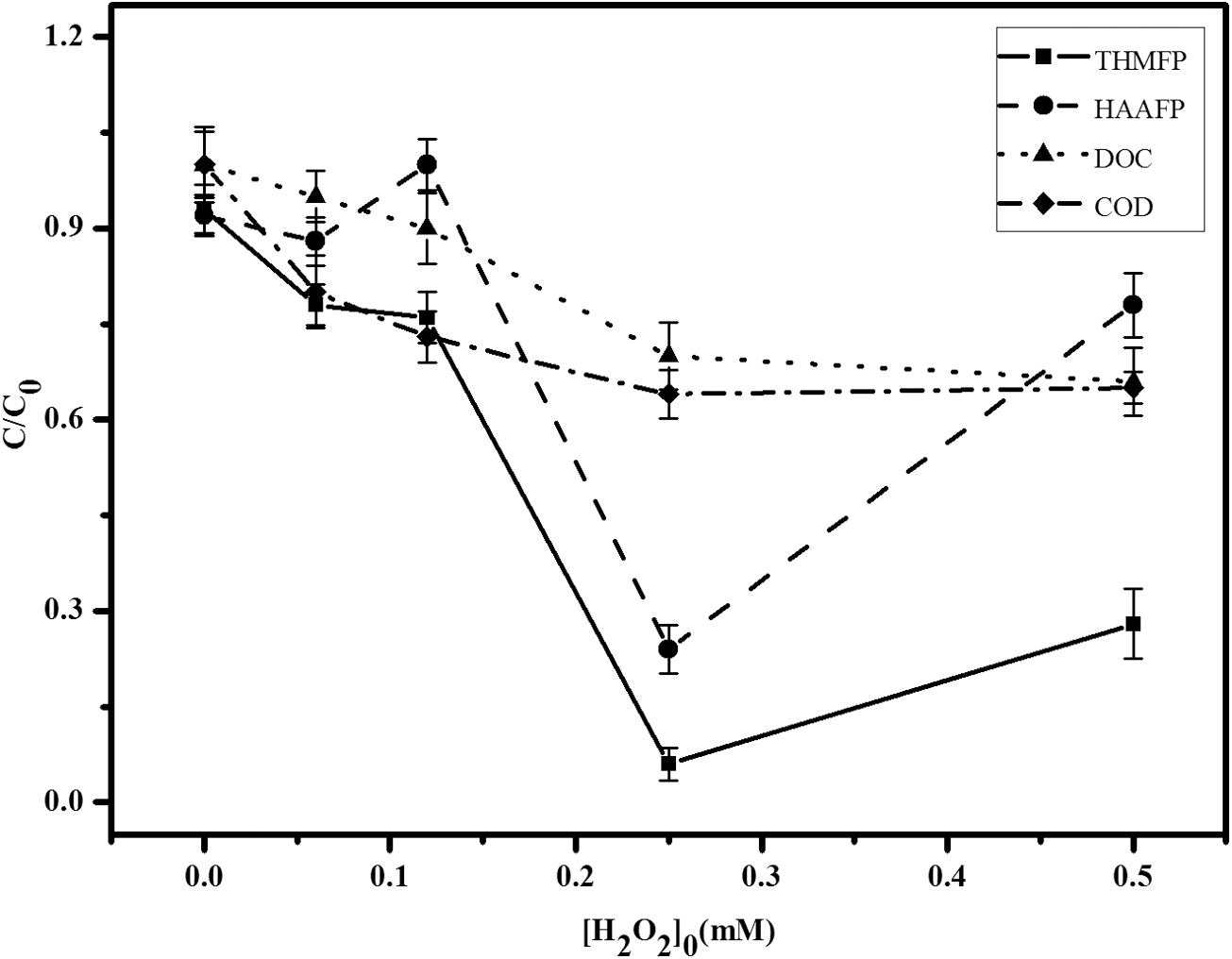
Effect of [H2O2]0 on measurements of THMFP, HAAFP, DOC, and COD after 90 min reaction time (Error bar indicates standard variation, the initial conditions: 2.0 mg/L DOC (resorcinol), 0.25 mM [Fe2+]0, and pH 5.0, the initial measurements (c0) of THMFP 3212 μg/L, HAAFP 84 μg/L, and COD 15.6 mg/L).
Effect of pH
When pH increased from 5.0 to 8.0, the concentrations of THMFP, HAAFP, DOC, and COD generally showed an increasing trend or the reductions by the oxidative reactions declined under the conditions of initial 2.0 mg/L DOC, 0.25 mM [Fe2+]0, and [H2O2]0 (Fig. 3). As pH was raised from 5.0 to 6.0, the reduction of THMFP dropped sharply from 94% to 46% and HAAFP from 77% to near zero. Meanwhile, the removals of DOC and COD were from 30% and 36% to 22% and 18%, respectively. At pH 7.0, THMFP remained relatively constant, similar to that at pH 6.0, while the removals of DOC and COD decreased further to 12% and 5%. Surprisingly, there was still about 24% reduction in HAAFP. At pH 8.0, there were no significant reductions in HAAFP or COD, but about 17% THMFP and 10% DOC reductions were still measured. Data demonstrated that acidic condition such as pH 5 could promote DOC removal and DBP control by the oxidative reactions.

Measurements of THMFP, HAAFP, DOC, and COD as affected by pH after 90 min reaction time (Error bar indicates the standard variation, the initial conditions: 2.0 mg/L DOC (resorcinol), 0.25 mM [Fe2+]0, and 0.25 mM [H2O2]0, the initial measurements (c0) of THMFP 3212 μg/L, HAAFP 84 μg/L, and COD 15.6 mg/L).
Effect of initial DOC
Three concentrations of initial DOC were selected and investigated to evaluate its impact on the reduction efficacy of THMFP and HAAFP. As shown in Fig. 4, a higher initial DOC would result in a higher reduction of either THMFP or HAAFP. At 2.0 mg/L DOC, the reductions of THMFP and HAAFP were 6% and 27% higher (95% and 77%), compared with those (88% and 50%) at 1.0 mg/L DOC, under the conditions of 0.25 mM of both [Fe2+]0 and [H2O2]0 and pH 5. When initial DOC was raised to 12.0 mg/L, the reductions of THMFP and HAAFP reached 98% and 92% under 1.0 mM [Fe2+]0 and [H2O2]0, pH 5 condition. In addition, 64% DOC and 60% COD reductions were also measured under the same condition (data not shown).
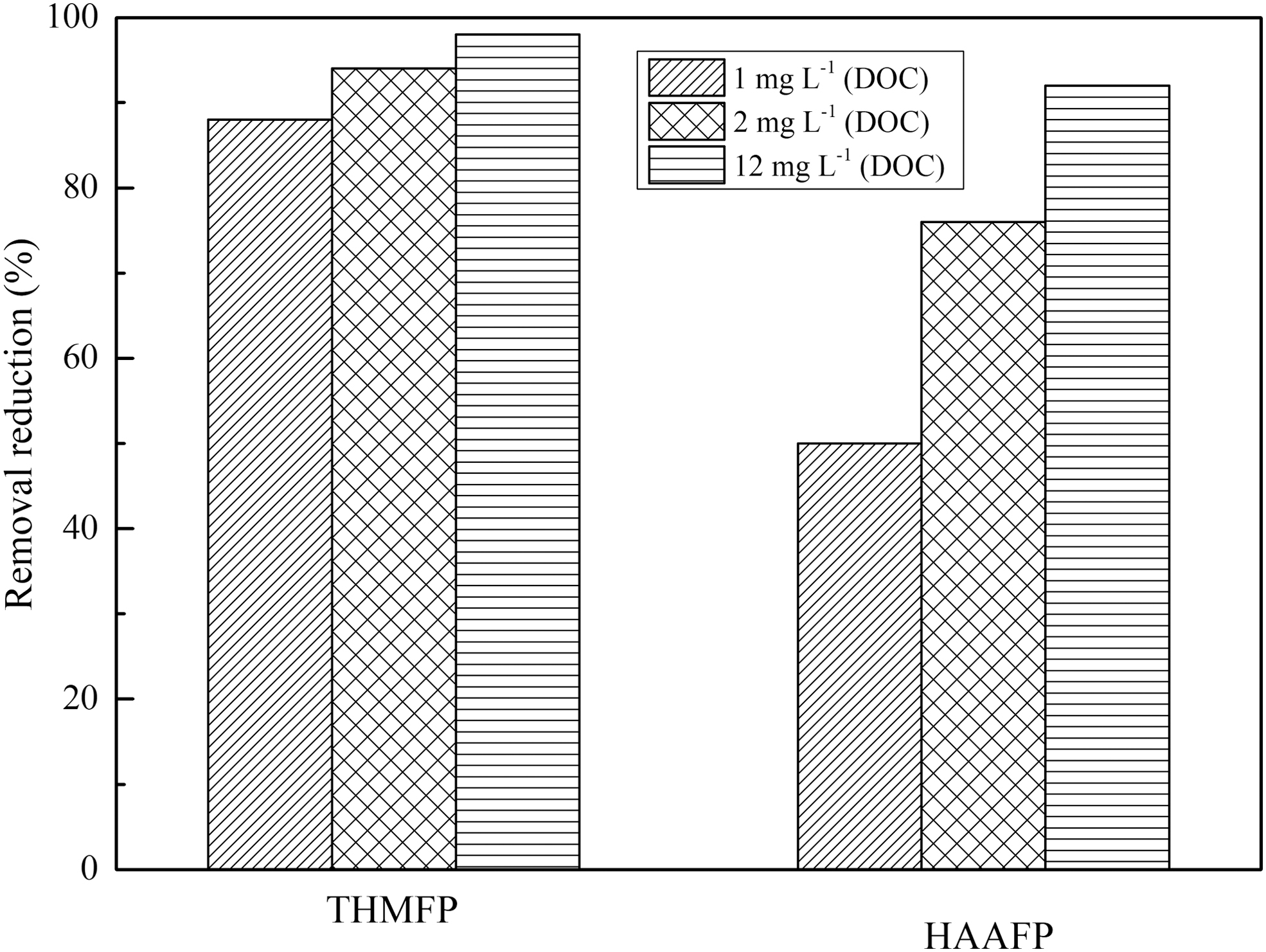
Removal efficacy of THMFP and HAAFP as affected by initial DOC concentration (Error bar indicates the standard variation, the initial condition: pH 5.0. For the tests under 1.0 and 2.0 mg/L DOC (resorcinol), 0.25 mM [Fe2+]0 and 0.25 mM [H2O2]0 were used. For the tests under 12.0 mg/L DOC (resorcinol), 1.0 mM [Fe2+]0 and 1.0 mM [H2O2]0 were used).
Competition of DOC removal and E. coli disinfection
In an effort to evaluate the competition of DOC removal with disinfection of microbial indicator E. coli, three sets of experiments were conducted, and the results are shown in Fig. 5. In the first experiments at the absence of DOC (resorcinol), no significant disinfection was detected with initial 7.2 × 105 CFU/mL E. coli under either 1.0 mM [Fe2+]0 or 1.0 mM [H2O2]0 only condition, as shown in the first and second bars in Fig. 5. In the second set of experiments, the initial reaction conditions were selected as 7.2 × 105 CFU/mL E. coli and 0.25 mM [Fe2+]0 and [H2O2]0. The results showed that almost 100% disinfection (<1 CFU/mL left) was achieved in the absence of DOC (data not shown), where in the presence of 2.0 mg/L DOC, there was still significant deactivation of E. coli, with only 30–40 CFU/mL E. coli left, as shown in the third (E. coli) bar in Fig. 5. However, no measurable DOC reduction was detected, as shown in the fourth (for DOC) bar in Fig. 5. The third set of experiments were conducted under the conditions of 7.2 × 105 CFU/mL E. coli and 1.0 mM [Fe2+]0, [H2O2]0, 12.0 mg/L DOC. The results showed that there was 100% disinfection achieved (data not shown) and 60% DOC removal, as shown in the fifth bar in Fig. 5, similar to the DOC removal efficiency in Fig. 4 for 12.0 mg/L DOC.
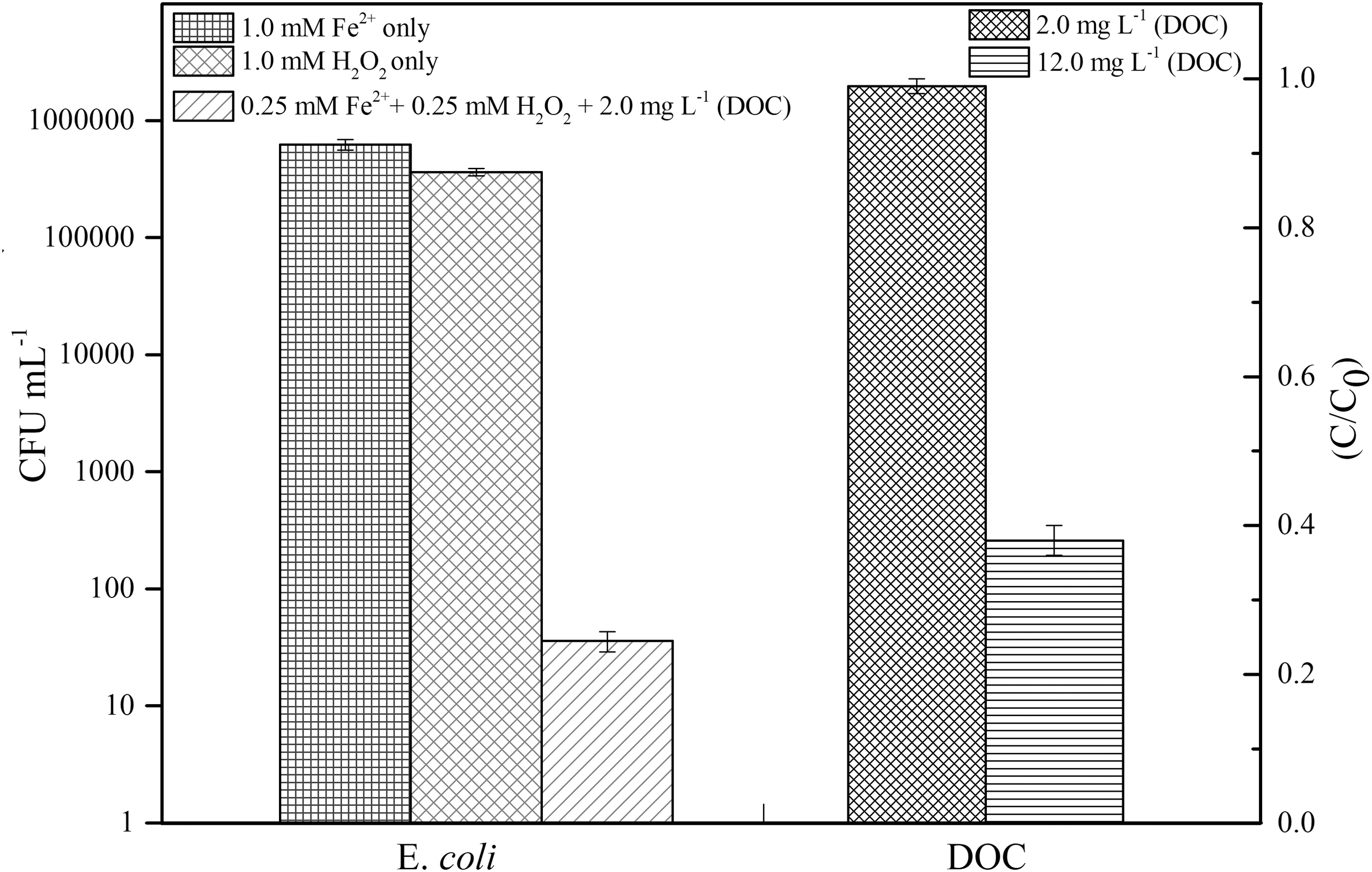
Comparison between DOC removal and Escherichia coli disinfection [Error bar indicates the standard variation. Reaction conditions: pH 5.0, First E. coli bar: initial 7.2 × 105 CFU/mL E. coli, 1.0 mM Fe2+ only; Second E. coli bar: initial 7.2 × 105 CFU/mL E. coli, 1.0 mM H2O2 only; Third E. coli and first DOC bars: initial 7.2 × 105 CFU/mL E. coli, 0.25 mM Fe2+ and H2O2, 2.0 mg/L DOC (resorcinol); Second DOC bar: initial 7.2 × 105 CFU/mL E. coli, 1.0 mM Fe2+ and H2O2, 12.0 mg/L DOC (resorcinol)]. CFU, colony forming units.
Discussion
Hydroxyl radicals (·OH), generated through the H2O2 decomposition catalyzed by ferric/ferrous ions, are stronger oxidative species than HClO generated by chlorination (Buxton et al., 1988). Theoretically, any moiety or molecule that can be oxidized by HClO should be also oxidized by hydroxyl radical as well, which would potentially lead to the reduction of the chlorinated oxidative DBP formation (e.g., THMs). Both Fenton (Fe2+/H2O2) and Fenton-like (Fe3+/H2O2) reactions have been explored for degrading a variety of organic contaminants in environmental applications. The mechanisms proposed are presented in Table 1 (Neyens and Baeyens, 2003).
Mechanisms of Fenton/Fenton-Like Reactions
Application of H2O2 during drinking water treatment operation may be of a public health concern. However, according to the U.S. National Sanitation Foundation Standard 60, H2O2 is a safe chemical in drinking water treatment, because its final decomposition products are harmless oxygen and water (NSF, 2019). H2O2 has been reported to being used for removing dissolved iron and excessive ozone in drinking water treatment. There are currently a few brands of H2O2 commercially available for drinking water treatment, for example, BIOX A, Oxy Pure.
In this study, resorcinol was selected as a surrogate for organic matter to mimic DOM in source water, because it had been well characterized in THMFP and HAAFP (Bond et al., 2012), with THMFP of 1500 ± 94 μg/mg C and HAAFP 58 μg/mg C. Our experiments illustrated that 3.1 mg of resorcinol generated 3212 ± 137 μg/L of THM and 84 μg/L HAA. As carbon mass ratio of 0.691 in resorcinol, 3.1 mg of resorcinol is equivalent to 2.1 mg C. Therefore, our experimental data were consistent with the values reported.
Our preliminary studies showed that only 10% of THMFP and 12% of HAAFP were reduced by 0.25 mM Fe3+/H2O2, with minimal declines of DOC or COD (data not shown), at the conditions of 2.0 mg/L DOC, pH 5.0, and 25°C ± 3°C, while 94% THMFP and 76% HAAFP reductions were achieved by 0.25 mM Fe2+/H2O2, as shown in Fig. 1. In comparison between Reactions 1 and 3 listed in Table 1, kinetic constant k3 was about three orders smaller than k1, generating much less ferrous ions for Fenton reactions [19]. As a result, Fe2+, instead of Fe3+, was selected to initiate Fenton reactions in this study.
As shown in Fig. 1, the reductions of DOC, COD, THMFP, and HAAFP were generally higher with increasing [Fe2+]0, but at the variable rates. From 0.0 to 0.25 mM [Fe2+]0, increased ferrous ions would generate more ·OH and enhance the oxidative processes, consequently leading to the higher reductions. The decline of DOC could be a result of advanced chemical oxidation or mineralization processes of resorcinol induced by Fenton reactions. Data indicated only 30% resorcinol mineralized and 36% resorcinol oxidized after the reactions. The reduction of THMFP or HAAFP could be attributed to (1) structure alteration of resorcinol or its oxidative byproducts that were much less reactive with chlorine; and (2) oxidation or mineralization of THM or HAA precursors. Further increasing [Fe2+]0 from 0.25 to 0.5 mM did not result in enhanced reductions of THMFP, HAAFP, DOC, or COD, which was not surprising. Reaction 5 and 6 in Table 1 indicated that Fe2+ could also act as a scavenger for reactive species (e.g., ·OH). As a result, 0.25 mM was the optimized [Fe2+]0 concentration for treatment.
The impacts of [H2O2]0 on the removal of DOC or COD were similar to those of [Fe2+]0, with a moderate enhancement as the reactant increased, but the change trend of THMFP or HAAFP was different compared with that of [Fe2+]0 (Fig. 2). As [H2O2]0 was raised from 0.0 to 0.25 mM, there was a fast reduction for both THMFP and HAAFP. However, when [H2O2]0 was from 0.25 to 0.5 mM, the reductions for both rebounded substantially. Although Reaction 1 in Table 1 showed that the generation rate of ·OH would increase with increasing [H2O2]0, Reaction 2 indicated that H2O2 was also a strong scavenger for ·OH generated. Therefore, excessive amount of H2O2 was not recommended as it would lower the treatment effectiveness in terms of DBP control, which could also lead to a higher treatment cost. A similar trend on the effects of increasing [H2O2]0 was previously reported (Murray and Parsons, 2004).
In general, a higher pH from 5.0 to 8.0 had a higher content or a lower removal efficiency of DOC, COD, THMFP, or HAAFP, as shown in Fig. 3. This observation could be linked to two attributes. First, alkaline pH was reported to favor the disproportionation of H2O2 to H2O and O2, rather than the generation of ·OH (Muruganandham and Swaminathan, 2004; Su et al., 2011). Second, a higher pH could promote the formation of amorphous ferric oxy(hydroxides) as shown in following reaction (Murray and Parsons, 2004), resulting in a faster deactivation of ferrous catalyst (Bigda, 1995). Therefore, the operation pH should be carefully monitored for practical application.
The reduction of THMFP or HAAFP by Fenton or Fenton-like reactions was similar regardless of initial DOC content, but disproportional to the concentration (Fig. 4). This trend suggested that the functional groups of DBP precursors that could be oxidized and reacted with chlorine for DBP formation were less than the amount of ·OH species generated. In addition, the ·OH species generated may be able to only partially oxidize DBP precursors or polymerize the intermediates formed during the reactions (Weber et al., 2005). The removal of DOC or COD could be also interpreted by the mechanisms mentioned above. This result indicated that the reactant dosage should be adjusted or optimized based upon the initial conditions of source water for a maximum treatment effectiveness and operational cost saving.
Iron species have been explored to inactivate or disinfect bacteria and viruses and shown that both Fe2+ and Fe3+ were effective to some degree (Kim et al., 2010; Spuhler et al., 2010; Ortega-Gómez et al., 2014). Data shown in Fig. 5 demonstrated that the oxidative species generated by Fenton reactions at 0.25 and 1.0 mM [Fe2+]0 and [H2O2]0 were able to significantly inactivate E. coli present, while neither Fe2+ nor H2O2 alone showed any significant disinfection effect. Results from the second set of experiments suggested that the competition for hydroxyl radicals between DBP precursors (resorcinol) and E. coli could affect disinfection efficiency and DBP control. In the third set of experiments, the concentrations of both reactants were raised to 1.0 mM to generate much more reactive species, where DOC was also raised to 12.0 mg/L to mimic river water. The results indicated that the reactive species generated by the reactions at 1.0 mM [Fe2+]0 and [H2O2]0 was sufficient to exhibit nearly the same removals toward THMFP and HAAFP mitigation with or without E. coli, while the reactive species generated at 0.25 mM [Fe2+]0 and [H2O2]0 in the second set of experiments may be insufficient for effective disinfection and DBP reduction simultaneously. Thus, in an effort to achieve a simultaneous disinfection and DBP mitigation, the reactant dosage should be adjusted and optimized based on the initial conditions of source water.
As our effort to develop a cost-effective drinking water treatment technology that would assist small, resource-limited water systems in compliance with the USEPA DBP regulations, this study has demonstrated a successful water treatment practice to enable control of DBP formation. However, deployment of this technology would heavily depend on operation costs. In general, it is a challenge to conduct a detailed cost analysis for the treatment operations, because operation costs are always affected by many factors, such as the characteristics of source water, reactant dosage, and so on. Several factors as following should be considered before implementation. First, if source water contains iron in mg/L level, adding less or no iron during treatment should be considered as an advantage. Many iron-containing compounds are also common coagulants in water treatment. The oxidation of ferrous to ferric iron by the reactions could be used in conventional coagulation/flocculation process for DOC removal in treatment (Murray and Parsons, 2004). Second, application of hydrogen peroxide, instead of chlorine, could potentially minimize the formation of chlorine-associated DBPs under slightly acidic (pH ∼5) condition, which is safe for drinking water usage. However, pH adjustment before and after the treatment is likely needed, depending on source water conditions. Third, simultaneous disinfection and DBP mitigation by the reactions could help lower chlorine dosage and associated costs. The cost saving from chlorine consumption, coagulation, and aeration may compensate for the costs associated with chemicals (H2O2, Fe2+, and acid) added, which would make this treatment potentially cost-effective to be successfully implemented in small water systems.
Conclusion
This study demonstrated that reductions of DOC, COD, THMFP, and HAAFP were dependent on pH, Fe2+, and H2O2 concentrations, as well as initial DOC content. The highly reactive oxidative species generated from the catalytic decomposition of hydrogen peroxide (0.25 mM H2O2) by ferrous (0.25 mM Fe2+) at pH ∼5.0 were able to reduce 94% THMFP and 77% HAAFP within 90 min. The oxidative reactions also resulted in 30% DOC and 36% COD reductions and achieved E. coli disinfection. The water treatment technology developed could be potentially used to replace the chlorine disinfection process in conventional drinking water treatment plant and help small, resource-limited water systems, as a green, cost-effective technology, in compliance with the USEPA DBP regulations and safeguard public health for rural residents through DBP mitigation and improved drinking water safety. Nevertheless, results identified that pH was one of the most important parameters affecting the efficacy of DBP control. The treatment would be more effective and economically efficient for relatively low pH or slightly acidic source water. Recarbonation could help lower source water pH and enhance DBP control efficacy. In addition, since resorcinol was used as a model DOC compound in this study to mimic DOM in source water, it should be mindful when the results are extrapolated to source waters containing diverse natural organic DBP precursors.
Footnotes
Acknowledgment
Authors thank Ms. Mary Reed for her technical assistance in E. coli measurement.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by USEPA and United States Department of Agriculture, National Institute of Foods and Agriculture (USDA-NIFA) through the grants (EPA no. 83517301 and NIFA no. 1004422 and 1026209) to Lincoln University of Missouri.