
Abstract
Fate of extracellular antibiotic resistance genes (eARGs)—which are widespread in various environments—is dependent on gene persistence. This work examined the previously unconsidered role of environmentally relevant nanoparticles (NPs) in regulating eARG persistence. Fragments of eARG blaI were added at 2 μg/mL to batch systems containing nuclease-free water at pH 7.0, 0.1 M CaCl2 and 0.25 g of particles. Tested particles included humic acid functionalized silica nanoparticles (HASNPs) and kaolinite. After equilibration of eARG fragments and particles, pH was changed (between 1.0 and 11.0) or DNase I was added (at concentrations between 0.5 and 20 μg/mL). Remaining eARG fragment copies were characterized by the quantitative polymerase chain reaction and automated gel electrophoresis. eARGs fragments of various sizes (508, 680, and 861 bp) and both double- and single-stranded eARGs were tested. Sorption capacity of DNase I to NPs was also assessed using the Bradford assay for protein detection. Overall, particles improved eARG persistence. HASNPs protected extracellular DNA (eDNA) from breakage in low pH systems. Kaolinite provided full protection at all DNase I concentrations tested. Degradation of eARGs was significant in HASNP systems but was markedly decreased compared with particle-free systems at DNase I concentrations <10 μg/mL. DNase I sorbed significantly to HASNPs. Full protection of eARGs was observed, at DNase I concentrations <5 μg/mL, when HASNPs were adsorbed to DNase I. The impact of HASNPs on reducing degradation (at 1 μg/mL DNase I) was observed for up to 4 h. Smaller and single-stranded fragments, which have greater sorption capacities to HASNPs, were not better protected. This study establishes the ability of naturally occurring NPs to decrease the degradation of eARGs, either through sequestration of eDNA or inactivation of nucleases by particles. The ability of NPs to increase eARG persistence may have implications for the dissemination of antibiotic resistance.
Introduction
One of the greatest current public health concerns is the global rise of antibiotic resistance. Effective risk assessment of antibiotic resistance spread requires monitoring the proliferation of antibiotic resistant bacteria (ARBs) and antibiotic resistance genes (ARGs) (Vikesland et al., 2017). ARBs and ARGs are known to be widespread across various environments. ARGs, including extracellular ARGs (eARGs), have been detected in freshwater (Wang et al., 2016; Guo et al., 2017), seawater (Bien et al., 2017), sediment and associated surface waters (Mao et al., 2014; Yuan et al., 2019), liquid animal wastes from animal feed operations (Pruden et al., 2006; McKinney et al., 2010), drinking water treatment plants and distribution centers (Pruden et al., 2006; Xi et al., 2009) and wastewater treatment plants (Rizzo et al., 2013; Zhang et al., 2018; Gardner et al., 2019; Lu et al., 2020). eARGs in such systems may transfer antibiotic resistance downstream to bacterial populations. Extracellular DNA (eDNA) has demonstrated the ability to transfer genetic information across microbial populations by natural transformation (Stewart and Sinigalliano, 1990; Paul et al., 1991; Baur et al., 1996; Nielsen et al., 1997; Zhu, 2006), as it can persist long enough to be incorporated into bacterial genomes (Bennett et al., 2004; Pruden et al., 2006; Gardner et al., 2019). This phenomenon has been demonstrated with eARGs as well (Poté et al., 2003). eARG dissemination may be a significant contributor to the global rise of antibiotic resistance.
Typically, eDNA in nature rapidly degrades owing to contact with ubiquitous extracellular bacterial nucleases, or DNases (Blum et al., 1997; Levy-Booth et al., 2007; Pietramellara et al., 2008). The association of extracellular genes with naturally occurring particles, however, may prevent degradation through nucleases and increase rates of genetic uptake. Several environmental particles—such as clays (Demanèche et al., 2001; Cai et al., 2006b; Pietramellara et al., 2007; Gardner and Gunsch, 2017; Chowdhury et al., 2021), sands (Slater et al., 2006), and humic and fulvic substances (Creccio and Stotzky, 1988; Saeki et al., 2010)—have demonstrated the ability to adsorb eDNA, including eARGs, to their surfaces. Degradability by DNase I has been demonstrated to decrease for eDNA bound to clays (Khanna and Stotzky, 1992; Paget et al., 1992; Demanèche et al., 2001; Cai et al., 2006a, 2007), humic acids (Creccio and Stotzky, 1988), and sands (Lorenz and Wackernagel, 1987) as compared with free eDNA (Harrison et al., 2019). This protection is suggested to be owing to either (1) conformational changes of eDNA induced by particle adsorption that prevent interaction with nucleases, (2) physical inaccessibility of bound eDNA to nucleases, or (3) sorption of nucleases to particles and subsequent enzymatic deactivation (Khanna and Stotzky, 1992; Cai et al., 2006a, 2007; Pietramellara et al., 2008). The addition of soil components in systems with DNase I has also been shown to increase bacterial natural transformation rates, because binding to particles protects eDNA from degradation before cellular uptake (Khanna and Stotzky, 1992; Levy-Booth et al., 2007; Pietramellara et al., 2007).
A particularly mobile fraction of particulate matter not yet evaluated for its role in regulating the persistence of eDNA or eARGs is made up of nanoparticles (NPs)—which are ubiquitous in natural environments and increasing in incidence from their use in anthropogenic applications (Peters et al., 2014; Norwack, 2016). NPs have demonstrated the ability to irreversibly sorb eARGs to their surfaces and alter their conformation (Chowdhury et al., 2021). The effect of NPs on eARG persistence may be important to study because nanoemergent traits—such as increased mobility (Wiesner et al., 2006) compared with micron-sized particles and ability to bioaccumulate in plants (Schwab et al., 2015)—may contribute to the spread of ARGs in yet unknown ways.
In this work, the persistence of eDNA, and in particular eARGs, bound to naturally occurring nanoparticles are explored. eARG fragments used are isolated from the widespread ARG blaI, which confers resistance to penicillins, cephamycins, and carbapenems by coding for a repressor protein of β-lactamase resistance genes (Pence et al., 2015). blaI is also an environmentally relevant ARG, as it was among the most commonly used selection markers in transgenic crops (Aeschbacher et al., 2005) and subsequently found in wastewaters (Jacobs and Chenia, 2007; Demaneche, 2008; Gardner et al., 2019). Particle-bound and free extracellular blaI fragments are exposed to varying pH conditions and concentrations of DNases in simplified model systems containing sterile nuclease-free water. After exposure, the remaining quantities of eDNA are evaluated. Particles used include pure silica nanospheres surface functionalized with humic acid and pure kaolinite. The nanospheres were selected as a simplified model nano-scale particle. They were surface functionalized with humic acid as this allows for significant attachment of the particle to eDNA (Chowdhury et al., 2021). The protective effect of kaolinite on eDNA has already been established in literature and is tested here as a basis to which the protective effect of the model nanoparticle can be compared. Persistence is also evaluated with eARG fragments of varying structural properties. eDNA's structural properties have demonstrated an impact on eARG sorption behavior (Chowdhury et al., 2021) and therefore are evaluated here to determine if sorption behavior correlates with eARG persistence. In sorption tests, the association of nucleases to NPs is also evaluated, as sequestering of nucleases by particles may impact eARG degradability.
The use of well-characterized materials—with mineral compositions often found in nature—provides a balance between environmental relevance and the simplification of a complex system with the goal of understanding mechanisms. Similarly, the use of DNases and sterile water allows the assessment of NPs' role in regulating eARG persistence without the further complexity of simultaneously considering the role of solution chemistry in these interactions. This work aims to perform an analysis of eDNA-particle interactions and impacts of these interactions on DNA persistence. This may allow for a broader understanding of how previously unconsidered naturally occurring NPs may influence the environmental fate of eARGs.
Materials and Methods
DNA
The research involves no more than minimal risk to subjects, therefore the requirement of approval by an Institutional Review Board was waived.
Model linear eDNA fragments (Table 1) were amplified through the polymerase chain reaction (PCR) from the ARG blaI. These exact fragments were prepared similarly as for a previous study (Chowdhury et al., 2021). PCR was performed using Hot Start Taq Polymerase reagents from New England Biolabs (Ipswich, MA) and custom DNA oligo primers (sequences in Table 1) from Integrated DNA Technologies (Coralville, IA). The PCR was conducted on the GeneAmp PCR System 9700 (Themo Fisher Scientific, Waltham, MA) at the following conditions: initial denaturation at 95°C for 30 s, followed by 35 cycles of 95°C for 30 s, 55°C for 60 s, and 68°C for 60 s, with an extension of 68°C for 5 min. Single-stranded DNA (ssDNA) was amplified using asymmetric PCR, which uses 50 × more forward primer than reverse primer in the PCR protocol for double-stranded DNA (dsDNA) (Heiat et al., 2016). PCR products were purified using the DNA Clean & Concentrator Kit from Genesee Scientific (San Diego, CA) and verified on a 1% agarose gel and Qubit 2.0 Fluorimeter (Thermo Fisher Scientific, Waltham, MA). The amplified fragments vary in size and strand conformation. These fragments were selected to evaluate the role of DNA properties in governing eDNA's protection by particles. These properties were of particular interest because they were shown to affect the nature of eDNA's association with particle surface (Chowdhury et al., 2021).
Extracellular Antibiotic Resistance Gene Fragments of Varying DNA Properties Used in Persistence Tests
ARG, antibiotic resistance gene; F, forward; PCR, polymerase chain reaction; qPCR, quantitative PCR; R, reverse.
Clay minerals and nanoparticles
Two model colloids were used in adsorption studies: kaolinite clay (Al2O3·2SiO2·2H2O) (Fluka Analytical, Charlotte, NC) and silica nanoparticles (Nanocomposix, San Diego, CA) surface functionalized with humic acid (HASNPs; Sigma-Aldrich, St Louis, MO). HASNPs were used as model NPs because the functionalization of environmentally relevant silica NPs with humic acid allowed for the adsorption of eDNA to particles (Chowdhury et al., 2021). HASNPs were prepared as described elsewhere (Chowdhury et al., 2021). All particles used were characterized for their hydrodynamic diameter and calculated zeta potentials. Details of particle size and surface charge are described in a previous study (Chowdhury et al., 2021). As the same exact particles were used as in a previous study, particles were not recharacterized. Measured hydrodynamic diameters were 865.73 ± 59.07 nm for kaolinite, 136.60 ± 1.29 for silica NPs, and 165.63 ± 23.64 for HASNPs.
Effect of pH on the persistence of eDNA
The persistence of eDNA in various pH conditions was tested in systems with HASNPs and in systems without particles. All tests were performed in 15 mL ultracentrifuge tubes. The tubes and particles were autoclaved before use to ensure the removal of any contaminants. Eleven pH values (1.0–11.0) were tested for each type of system, and samples at each pH were tested in triplicate. Although extreme pH values are less environmentally relevant, they were tested to thoroughly characterize any observable trends in particles' protective effect on eDNA. All tests were performed at 20°C.
For tests with particles, 2 μg/mL of eDNA (fragment 3, Table 1) was mixed into 500 μL solutions of 1 mM CaCl2 with 0.25 g of HASNPs. Fragment 3 was used because it had previously shown the highest adsorption capacities to the tested particles, which may allow for observable particle effects to be more pronounced in systems with varying pH. CaCl2 was used because divalent ions were shown to facilitate sorption of eDNA to particle surfaces (Chowdhury et al., 2021). This cation concentration was chosen because it is within realistic environmental limits. The CaCl2 solution was made using molecular biology grade sterile water from New England Biolabs, which has been tested to be free of endotoxins, nuclease activity, RNase activity, or any material detectable by a ultraviolet-visible scan between 200 and 800 nm. This mixture was adjusted to pH 7.0 by the addition of either HCl or NaOH and shaken (1 h, 80 rpm). This was performed to allow particles to first equilibrate with and sorb to eDNA at neutral pH. Then, pH was adjusted to values between 1.0 and 11.0. The mixture was shaken again (1 h, 80 rpm). This was performed to expose particle-bound eARGs to various pH conditions. For the negative control, the mixture was maintained at pH 7.0 for both hours of shaking. The mixture was then centrifuged (20 min, 20,000 g) and the supernatant was collected for analysis. The particle pellet was washed twice with 10 μM Tris buffer (pH 7.0) (which has demonstrated the ability to desorb loosely bound eDNA) (Cai et al., 2006b; Chowdhury et al., 2021), vortexed (3 min, 1,200 rpm), and centrifuged (20 min, 40,000 g). The supernatants were collected to capture any desorbed eDNA. All supernatants were washed with the One-Step PCR Inhibitor Removal Kit from Genesee Scientific to remove excess humic acid. Samples were stored at −20°C before downstream analysis.
For tests without particles, eDNA was added to 500 μL of 1 mM CaCl2 at pH 7.0 and the mixture was adjusted to pH values between 1.0 and 11.0. The pH was altered from neutral immediately in particle-free tests because eARGs were the only substance in solution and did not need time to equilibrate or adsorb to surfaces. The mixture was shaken (1 h, 80 rpm) and stored at −20°C for downstream analysis.
Persistence of DNase I-exposed eDNA
eDNA (fragment 1, Table 1) was exposed to DNase I in systems with (1) kaolinite, (2) HASNPs, and (3) no particles. Five different DNase I concentrations (0.5–20 μg/mL) were tested for each type of system, and samples at each concentration were tested in triplicate. Fragment 1 was used in these tests because it has demonstrated the capacity to adsorb to both particles (Chowdhury et al., 2021) and it is the longest gene fragment. The length may be useful in characterizing the extent of DNA fragmentation. All tests were performed at 20°C.
For tests with HASNPs or kaolinite, 2 μg/mL of eDNA was mixed into solutions of 1 mM CaCl2 with 0.25 g of HASNPs and adjusted to pH 7.0. This mixture was shaken (1 h, 80 rpm). Then, various volumes of bovine pancreas DNase I (Sigma-Aldrich) were added to achieve five different DNase I concentrations between 0.5 and 20 μg/mL, with a total liquid volume of 500 μL. The mixture was shaken again (1 h, 80 rpm). For the negative control, no nucleases were added during both hours of shaking. The mixture was then centrifuged, the pellet was washed, and supernatants were collected as described previously for pH tests in particle systems. This set of tests was also repeated using 0.25 g kaolinite clay in place of HASNPs.
For tests without particles, eDNA was added to 1 mM CaCl2 at pH 7.0 and the mixture was shaken for 1 h (80 rpm). It was adjusted by the addition of DNase I at five different concentrations between 0.5 and 20 μg/mL, up to a total liquid volume of 500 μL. The mixture was shaken for another hour (80 rpm) and collected for downstream analysis as described previously.
The remaining tests were performed with either HASNPs or without particles. After comparing the protective effect of HASNPs to kaolinite in initial tests, the already well-researched protective effect of kaolinite was not further evaluated in this study. Tests were repeated for fragments 2–4 (Table 1) to determine the impact of DNA properties (fragment length and strand conformation) on eDNA's persistence. All fragments were tested in HASNP and particle-free systems, in triplicate at each of the five DNase I concentrations (0.5–20 μg/mL). All tests were performed at pH 7.0 and 20°C.
The kinetics of eDNA degradation by DNase I was also examined. For kinetics tests, eDNA (fragment 1, Table 1) was shaken in 1 mM CaCl2 at pH 7.0 and 20°C for 1 h, with HASNPs or no particles. DNase I was added at a concentration of 1 μg/mL, in a total liquid volume of 500 μL. This mixture was then shaken for five time points between 15 min and 4 h. Samples at each time point were collected in triplicate. The samples were collected for downstream analysis as described previously for particle and particle-free systems.
Tests were also performed to determine whether the adsorption of DNase I onto particle surfaces impacts degradability of eDNA. To perform tests with particle-bound DNase I, DNase I was added to HASNP mixtures in 1 mM CaCl2 at pH 7.0 and 20°C at five concentrations between 0.5 and 20 μg/mL, up to a total volume of 500 μL. The mixture was shaken (1 h, 80 rpm) and centrifuged (20,000 g, 20 min). The supernatant was collected and the pellet of particles and adsorbed DNase I was then resuspended in 1 mM CaCl2. eDNA (fragment 1, Table 1) was then added at a concentration of 2 μg/mL, up to a total volume of 500 μL, and shaken again (1 h, 80 rpm). The mixture was centrifuged, the pellet was washed, and supernatants were collected as described for previous tests in particle systems. The results were compared with those from tests performed with free DNase I, as described previously.
Adsorption of DNase I
To evaluate the adsorption capacity of DNase I to particles, DNase I was added at four concentrations between 5 and 80 μg/mL to 0.25 g HASNPs in molecular biology grade sterile water at a total volume of 500 μL. The solutions were held constant at pH 7.0 and 20°C. Samples at each concentration were tested in triplicate. The mixture was shaken (2 h, 80 rpm) and centrifuged (20 min, 20,000 g). The supernatant was analyzed for DNase I concentration through the Bradford assay for protein quantification. To perform the Bradford assay, supernatants were combined with equal volumes of Bradford Dye (Bio-Rad Laboratories, Hercules, CA), vortexed, and read on the BMG SpectroStar Nano (BMG Labtech, Cary, NC) for absorbance at 595 nm. Absorbance was related to DNase I concentration based on a standard curve developed for DNase I with Bradford dye (Supplementary Fig. S1). The difference between initial and final DNase I concentration in the supernatant was taken to determine the amount of adsorption.
Analysis of DNA degradation
Free and desorbed eDNA in supernatants from each test were combined and DNA mass was quantified by the Qubit Fluorometer (Thermo Fisher Scientific). DNA copy number in each sample was determined by quantitative PCR (qPCR). For qPCR, each blaI fragment was amplified using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories). All reaction wells were prepared with 25 μL volume including (1) 13 μL of molecular biology grade sterile water; (2) 10 μL of master mix; (3) 1 μL each of forward and reverse primers; and (4) 1 μL of template DNA. Assays were run on the Bio-Rad CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) under the following conditions: initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 45 s, 55°C for 60 s, and 72°C for 30 s. Standard curves and no template controls were generated for each analysis. All samples were analyzed in triplicate. Primers used for each fragment were the same as those used for PCR (Table 1) and sample standard curves are given in Supplementary Fig. S2.
The quality of eDNA postexposure was assessed by the Duke University Microbiome Shared Resource by running 1 μL of each collected sample on the Agilent 2100 Bioanalyzer, which uses microfluidics technology to determine eDNA fragment sizes and concentrations through automated gel electrophoresis. Sample gels are given in Supplementary Fig. S3. The range of eDNA fragment sizes demonstrate the extent of fragment breakage postexposure.
Statistical analysis
All data points were collected in triplicate and standard error of the mean was determined. Copy numbers of DNA as determined by qPCR, N, were reported normalized to DNA copy numbers in the negative controls for each respective test, N0. The significance of particle addition and/or DNA properties was determined by analysis of variance (ANOVA). Significance was assumed at p < 0.05.
Results and Discussion
Persistence of eDNA in extreme pH
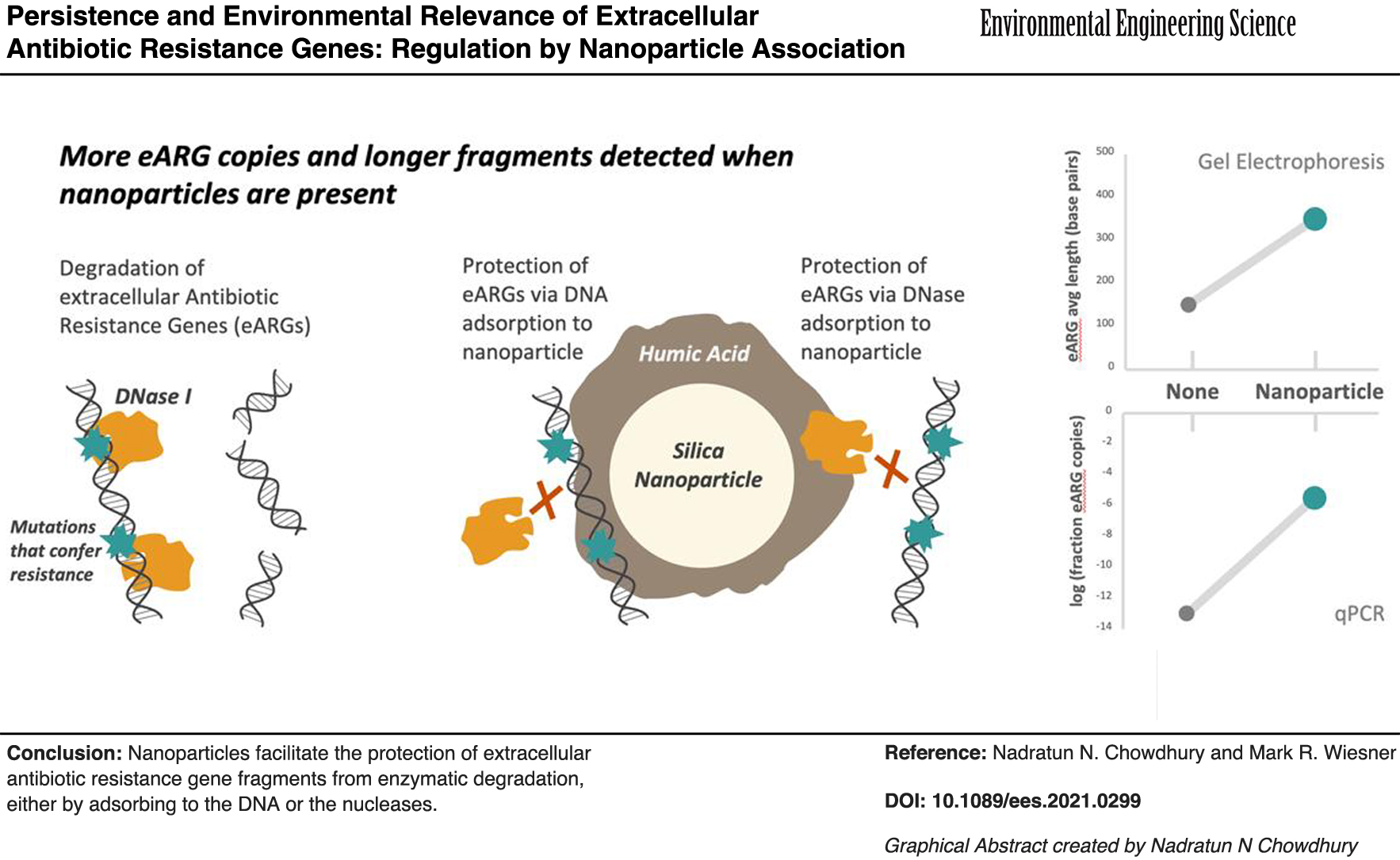
eARGs remained persistent across an environmentally relevant pH range (Fig. 1A). No statistically significant effect of adding HASNPs was observed (p < 0.70). eDNA degradation was only observed at pH 1, at which eARGs were completely degraded in particle-free systems, but short fragments were observed in HASNP systems (Fig. 1B). Binding of eDNA to HASNPs has been proposed to include bonds between humic acid's phenolate groups and eDNA's phosphate groups (Saeki et al., 2010). At acidic pH, humic acids have been shown to have a more condensed conformation (Prado et al., 2011). eARGs bound to compressed humic acids may have been less susceptible to acid-catalyzed hydrolysis.
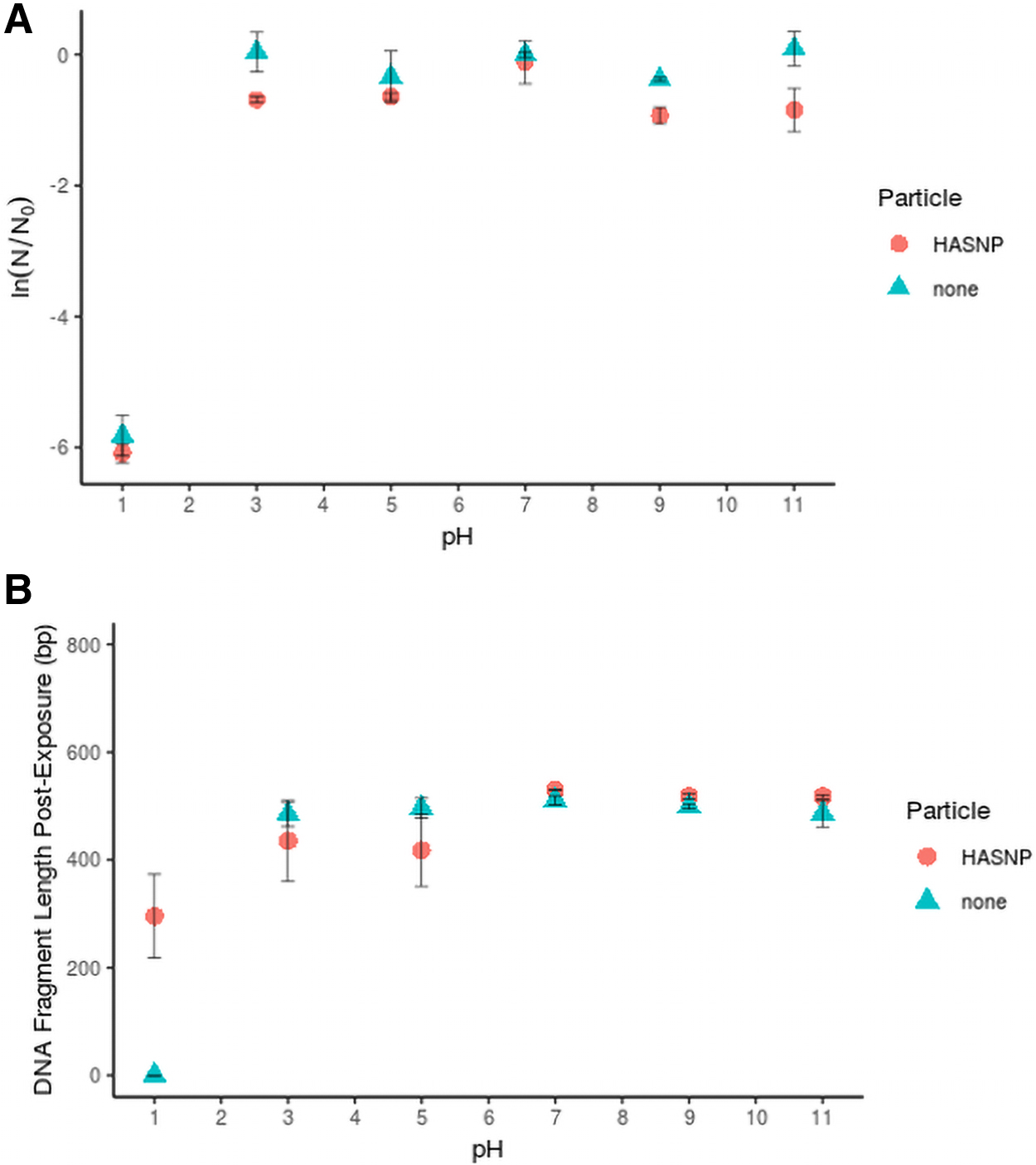
Impact of HASNPs on—
Persistence of DNase I-exposed eDNA
Particulate phases decreased the extent of eARG degradation by nucleases. A significant effect of particle addition and particle type on normalized DNA copy number was observed (p < 0.00072) (Fig. 2A). This protective effect may be explained by various hypotheses. It has been suggested that sorption of eDNA, particularly to humic substances, may render eDNA physically inaccessible to nuclease binding sites (Pietramellara et al., 2008). It has also been reported that eDNA fragments change their electron distribution and conformation when bound to certain particles. This change of bound eDNA conformation has been demonstrated by FTIR analysis (Cai et al., 2006a), including for eARGs bound to HASNPs (Chowdhury et al., 2021). These conformational changes may have caused eDNA's inability to interact with nuclease binding sites. Another hypothesis for the mechanism of the particles' protective effect is that the particles may have sorbed to the nucleases and become enzymatically inactivated (Khanna and Stotzky, 1992; Cai et al., 2006a).

Impact of particles on—
In kaolinite systems, a majority of eARGs remained persistent, even in systems with the highest DNase I concentrations tested. This suggests that DNase I was either deactivated by kaolinite, or that all eARG fragments were sorbed to kaolinite and unavailable to DNase I. In HASNP systems, a considerable degree of degradation was observed, but significantly greater copy numbers of eARG fragments were detected relative to particle-free systems, when DNase I concentration was up to 10 μg/mL. This indicates that the environmentally relevant NP can decrease enzymatic degradation of eDNA. However, this effect is much less pronounced than that by the well-characterized micron-sized clay, kaolinite. Because eARGs still degrade by several orders of magnitude in HASNP systems, it is unclear whether HASNPs protect eARGs to a sufficient extent as to increase their environmental relevance. It is reasonable to suggest, however, that eARGs bound to HASNPs may be more environmentally relevant than free eARGs, which are much more easily degraded.
Differences between the protective capacity of kaolinite and HASNPs may be owing to differences in particle composition and their influence on eDNA interaction. Sorption to kaolinite changes eDNA conformation differently than sorption to other particles. This may possibly be owing to the formation of strong bonds between kaolinite and eDNA that subsequently leads to distortion of molecular structure (Paget and Simonet, 1994; Cai et al., 2006a). Furthermore, kaolinite's comparatively stronger protective effect may correlate with its higher sorption capacity to eARGs as compared with HASNPs (Chowdhury et al., 2021). If kaolinite has a greater adsorption capacity than HASNPs for DNase I as well, this may also contribute to kaolinite's greater protective effect.
Breakage of the 861 bp eARG fragments was observed in all conditions (Fig. 2B). Excluding the highest DNase I concentration tested, there was a statistically significant effect of particle addition on eARG fragment length postexposure (p < 0.039). It seems to be a contradiction that DNA copy numbers remained essentially constant in kaolinite systems at all DNase I concentrations tested (Fig. 2A), but the average size of the fragments in kaolinite system appeared to decrease with the addition of DNase I (Fig. 2B). On kaolinite, eDNA is most likely to bind to −OH groups on the outer surface of the clay's octahedral sheets or Al3+ sites on the edges of the clay lattice (Poly et al., 2000; Cai et al., 2006b), from which it may extend outward and be more potentially accessible to nucleases. The portion of eDNA extending outward from the clay may have been prone to breakage, whereas the portion that forms strong bonds with kaolinite was not. Fragmented portions eDNA may have been detected as copy numbers by qPCR if even one of the primers was able to attach to them and amplify a single strand. Although fragmentation of amplicons would be expected to decrease the number of DNA copies detected by qPCR, significant amplifications of single strands may have occurred to detect relatively high copy numbers.
Although less eDNA adsorbs to HASNPs than kaolinite, HASNP-bound eDNA may have been better protected from breakage. It has previously been suggested that eDNA may penetrate into three-dimensional netted macropores formed on the surface of organic clays and be shielded from access by nucleases (Cai et al., 2006a). A similar phenomenon may occur on the surface of the HASNPs, which are coated with humic acid. Spaces within humic acid molecules have been characterized to be up to 15.762 nm (Yin et al., 2014), which may be large enough to fit eDNA fragments. eARGs have also demonstrated a unique compressed conformation when bound to HASNPs compared with kaolinite (Chowdhury et al., 2021), and this compression may lead to decreased breakage.
At higher DNase I concentrations, the nucleases were able to degrade eDNA fragments bound to each particle to the same extent as free eDNA. The highest DNase I concentration tested was not enough to completely disintegrate eDNA, as fragments were observed in all cases. HASNPs provided more protection to eDNA relative to the particle-free condition for all tested fragment sizes, particularly at lower DNase I concentrations (Fig. 3). A statistically significant effect of adding HASNPs was observed (p < 0.0097). There was no significant difference determined in the protection capacity of HASNPs between fragments of different sizes, measured both in ln(N/N0) (p < 0.707) and the fraction of remaining eDNA mass, M, to the mass of eDNA in the negative control, M0, as determined by the Qubit (p < 0.96) (data not shown). Protection capacity did not significantly differ between eARG fragments of various sizes even if data collected at the highest two DNase I concentrations tested, which appear to indiscriminately degrade all eARG fragments, were excluded from statistical analysis (p < 0.46). Although eARG fragment size was shown to significantly affect sorption capacity of eARGs to HASNPs (Chowdhury et al., 2021), the better sorbed smaller fragments were not more resistant to DNase I degradation. This indicates that factors other than eDNA sorption to the particle surface may be governing the ability of particles to protect eDNA from DNases.

Protection of HASNP-bound dsDNA of varying fragment lengths (dsDNA from blaI, fragments 1–3) from DNase I degradation at pH 7.0 and 20°C.
Both ssDNA and dsDNA showed less degradation by DNase I in HASNP systems compared with particle-free systems (Fig. 4). A statistically significant effect of adding HASNPs was observed for both (p < 0.013). There was no significant difference determined in the protection capacity of HASNPs between ssDNA and dsDNA, measured both in ln(N/N0) (p < 0.23) and M/M0 (p < 0.97). Strand conformation did not significantly impact protection capacity of HASNPs even if highest two DNase I concentrations tested were discarded from analysis (p < 0.23). Although ssDNA has been shown to have a higher sorption capacity to HASNPs than dsDNA (Chowdhury et al., 2021), it was not better protected from nuclease degradation than dsDNA. ssDNA is likely less stable than dsDNA and is degraded more easily than dsDNA (Gardner and Gunsch, 2017). ssDNA also desorbs more easily from the surface of HASNPs than dsDNA (Chowdhury et al., 2021), and therefore a greater fraction of free ssDNA may be available to DNase I than free dsDNA.
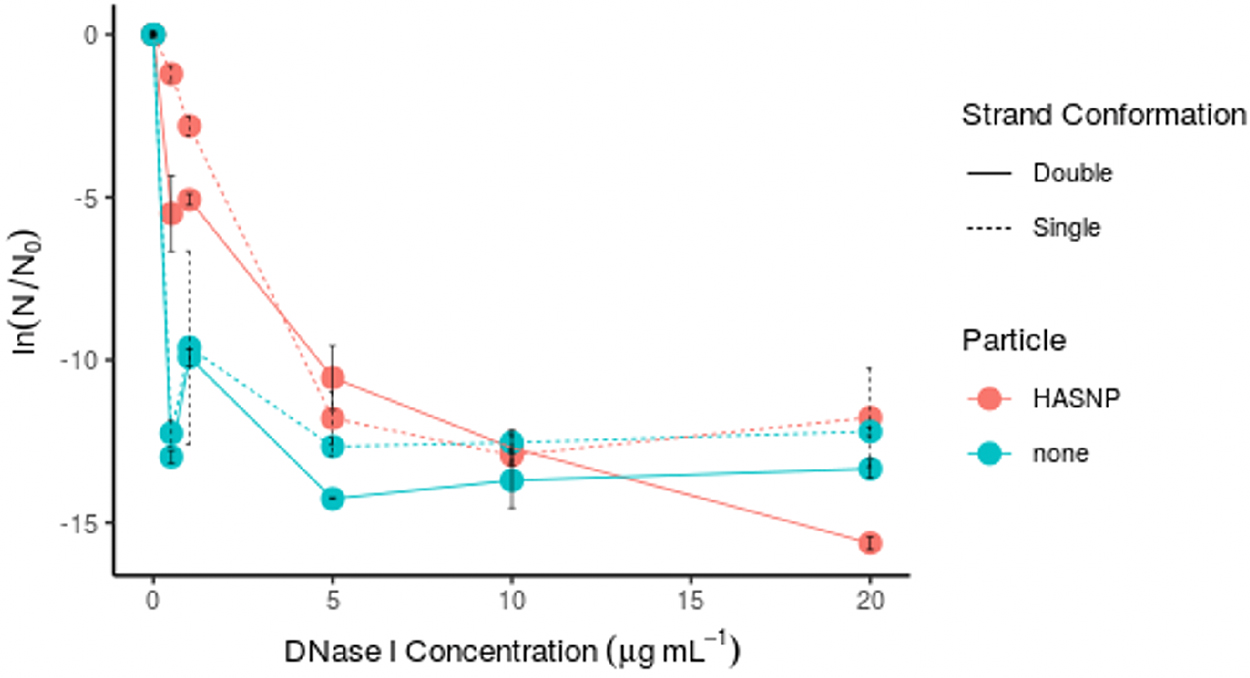
Protection of HASNP-bound ssDNA (861 bp ssDNA from blaI, fragment 4) from DNase I degradation at pH 7.0 and 20°C. ssDNA, single-stranded DNA.
DNase I adsorption capacity
DNase I adsorbed to the surface of HASNPs to its full capacity (Fig. 5). Adsorption capacity was assumed to be met when the adsorbed DNA concentration on the particle surface at equilibrium, qe, stopped increasing even as the equilibrium DNA concentration in solution, ce, continued to increase. Reaching this plateau suggests that the particle's binding sites are saturated and can no longer bind substrates. The adsorption data were fit to the linearized Langmuir adsorption model (Supplementary Text S1 and Supplementary Fig. S4 and Supplementary Table S1). The maximum adsorption capacity for DNase I, estimated by fitting to the Langmuir model, was ∼54 μg/g. This indicates that most of DNase I added in subsequent persistence tests was likely to have adsorbed to particles.
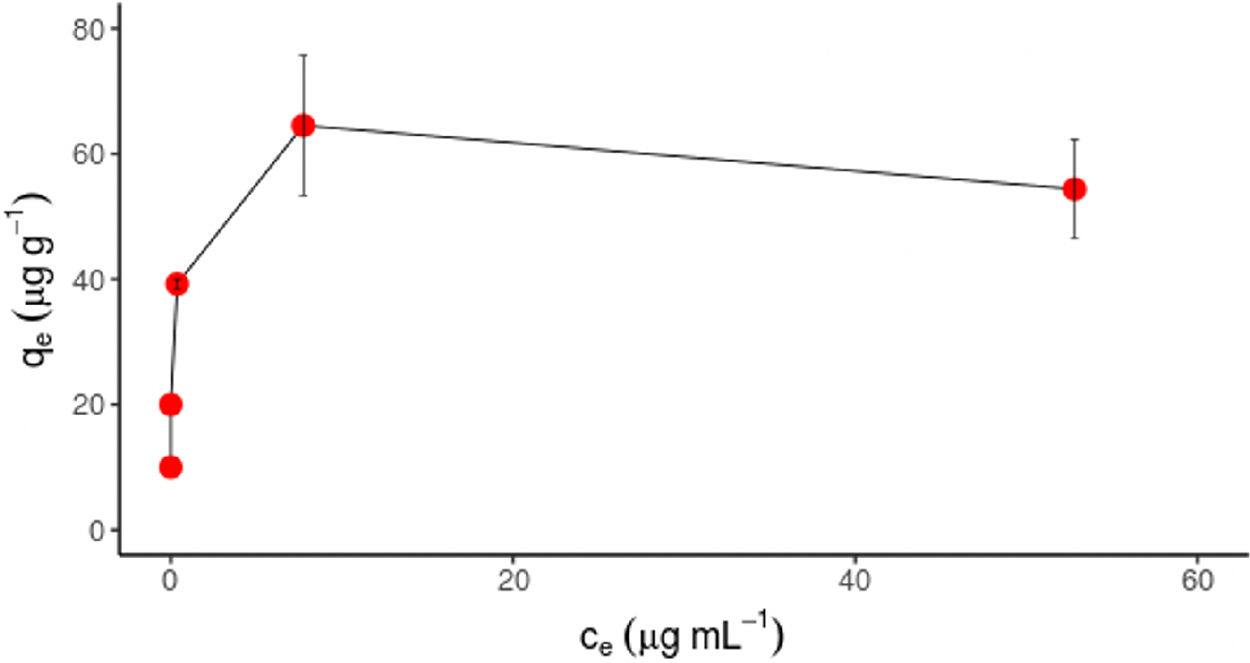
Adsorption isotherm of DNase I to HASNPs at pH 7.0 and 20°C.
Persistence of eDNA exposed to particle-bound DNase I
HASNPs provided some degree of protection to eARGs exposed to low DNase I concentrations, both in systems with free DNase I and adsorbed DNase I (Fig. 6). There was no statistically significant effect of DNase I adsorption observed overall (p < 0.34), but there was a significant effect of DNase I adsorption at concentrations below 10 μg/mL (p < 0.019). At the two lowest DNase I concentrations tested, the systems with HASNP adsorbed DNase I appeared to provide nearly full protection to eARGs, as the observed N/N0 ratio was close to 1. This full protection is not observed in HASNP systems with free DNase I. This suggests that DNase I, at these low concentrations, gets completely deactivated by adsorption to particles. It was hypothesized that enzymatic activity would be reduced not only by eDNA's association with particles, but DNases' association with particles. Both hypotheses appear to be supported by the data. This finding is supported by work in previous studies that have demonstrated the reduced activity of DNase I bound to a variety of environmental clay particles as compared with free DNase I (Khanna and Stotzky, 1992; Cai et al., 2006a).
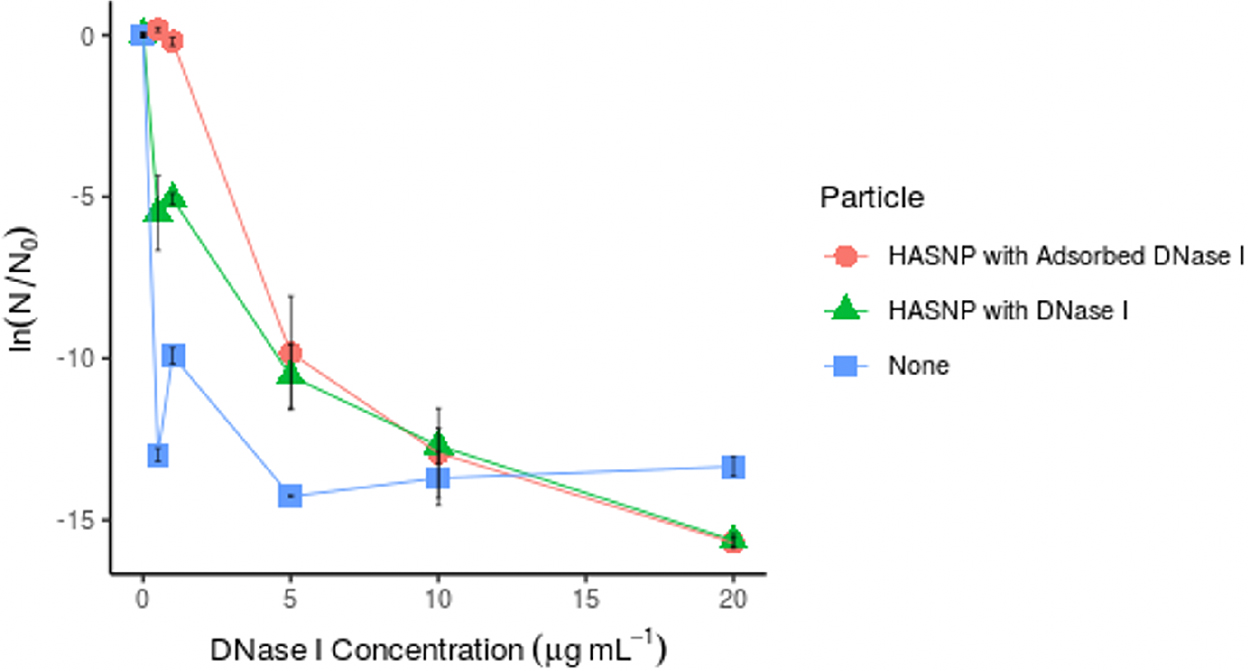
Protection of extracellular antibiotic resistance gene fragments (861 bp dsDNA from blaI, fragment 1) by DNase I adsorption to HASNPs measured in normalized gene fragment copy numbers (N/N0), at pH 7.0 and 20°C.
When DNase I concentration was increased to 5 μg/mL, the adsorption of DNase I to HASNPs no longer appeared to increase eARG protection, as N/N0 values are similar for free and adsorbed DNase I conditions. In this case, HASNPs appear to provide only slight protection to eARGs relative to the particle-free condition. At the higher DNase I concentrations tested, the DNase I activity was great enough to degrade eARGs significantly in all experimental conditions. When DNase I concentrations increase, the reduction of DNase I activity by particle sorption may be less relevant.
Kinetics of eDNA degradation
DNase I degradation occurred relatively quickly. Both HASNP and particle-free systems reached equilibrium within 1 h (Fig. 7). In the particle-free test, free eDNA remained in the system for its entire duration, so DNase I was likely the reaction-limiting agent. DNase I binding sites may have gotten saturated and stopped degrading eDNA within 1 h. The copy numbers of eDNA remaining at equilibrium were higher in HASNP systems than particle-free systems. The amount of available free eDNA may have been the reaction limiting reagent in HASNP system, as DNase I activity was less than what was observed in the particle-free system.
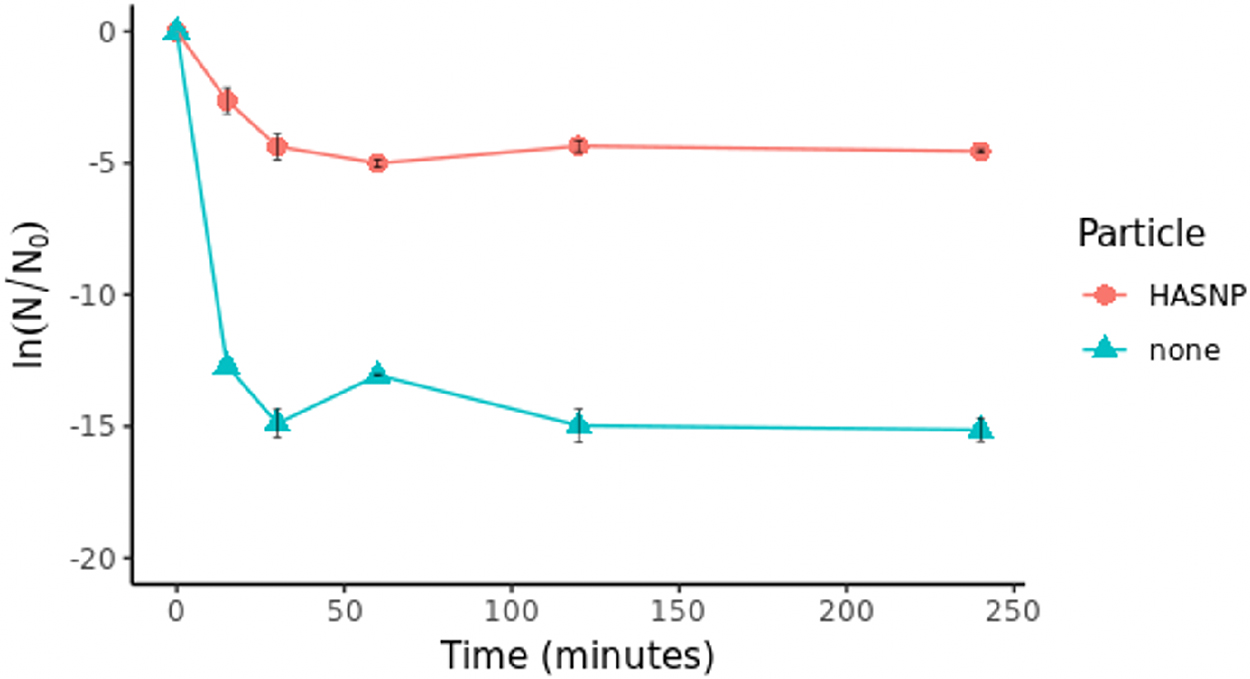
Kinetics of DNase I degradation of HASNP-bound DNA (861 bp dsDNA from blaI, fragment 1) measured in normalized gene fragment copy number (N/N0).
Up to 4 h of incubation did not show a drop in the remaining DNA copy numbers in HASNP systems, indicating that HASNP can protect eDNA over significant periods of time at the low DNase I concentration tested (1 μg/mL). This duration and extent of protection may decrease with increasing DNase I concentrations (i.e., >5 μg/mL), because higher DNase I concentrations lead to more eARG degradation in HASNP systems, as demonstrated in Figs. 2–4 and 6.
Although there was a difference in total DNase I activity, there was no statistically significant difference between the rate of DNA degradation in particle and particle-free systems (p < 0.47). This suggests that in the first hour or so, eARGs and DNase I interact with each other in bulk solution and HASNPs do not influence the rate of the reaction. HASNPs may have played a role in deactivating some of the enzyme or sequestering some of the eDNA by sorption, but not to the extent that it would affect the rates at which eDNA interacted with nucleases.
Conclusion
The results of this study imply that extracellular genetic material, including released ARGs, may persist in the environment for significant periods of time in association with more particles than previously reported. NP-associated eARG fragments were better protected relative to free eARGs (at DNase I exposures <10 μg/mL). The adsorption of DNase I to the HASNP surface enhances the protective effect and allows for full protection of eARG fragments (when DNase I concentration is <5 μg/mL). Therefore, it can be generally speculated that NPs coated with natural organic matter may enhance the persistence of environmental genetic materials. DNA properties did not appear to influence the persistence of eARGs, indicating that gene fragments from various sources could be equally susceptible to the effects of NP interaction.
These results imply that NP-associated eARGs may be better protected from enzymatic degradation than free eARGs in the environment. It is possible that this effect may increase the bioavailability of eARGs for bacterial uptake. It is also possible that NPs could keep eARGs sequestered from bacterial uptake if NP-bound eARGs are less bioavailable or suspended in transport. Further study is required to determine whether the degree of protection provided by HASNPs is sufficient to observe effects in eARG uptake or spread. NP effects on extracellular genes, although may be relevant, have not yet been monitored or accounted for in the characterization of antibiotic resistance spread.
Although this study provided initial confirmation of NP-mediated protection of eARGs from enzymatic degradation, its implications are limited by simplifications to complex environments imposed by the use of a model system. Real environmental systems include many components that could influence the ability of NPs to protect eARGs. Further study is required to characterize NP-eDNA interactions in realistic environments and elucidate their contribution to the rise of antibiotic resistance.
Footnotes
Authors' Contributions
N.N.C.: conceptualization, methodology, software, validation, formal analysis, investigation, writing—original draft, writing—reviewing and editing, visualization.
M.R.W.: conceptualization, writing—reviewing and editing, supervision, funding acquisition.
Acknowledgments
This work was aided by the services of the Duke University Microbiome Core Facility at the Duke Center for Genomic and Computational Biology, which performed automated gel electrophoresis on experimental samples. The development and validation of research ideas was also aided by Duke University professors Dr. Claudia Gunsch and Dr. Joshua Granek.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This material is based upon work supported by the National Science Foundation GRFP 1644868. This work was also partially funded through the Center for the Environmental Implications of NanoTechnology (CEINT) under NSF Cooperative agreement number EF-0830093.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.