
Abstract
Wildfires have become increasingly common across the United States in recent decades, with significant implications for ecosystem health and sustainability. The viability and metabolic activity of soil bacteria are key for maintaining global nitrogen-cycling processes and are likely to be impacted as burn severity continues to increase. To this end, wildfire-affected soils were studied to examine the impact on nitrogen-cycling bacteria in soils affected by low, moderate, and high wildfire severities. This objective was achieved by characterizing soil bacterial communities in control (i.e., unburned) and burned soils collected one year following the Woolsey wildfire (Los Angeles and Ventura Counties, CA, USA) using Illumina MiSeq 16S sequencing and profiling nitrogen-cycling gene expression using quantitative reverse transcription polymerase chain reaction (qRT-PCR). Six families and 17 genera were significantly (Spearman rs > |0.4|; p < 0.05) negatively associated with wildfire severity, and three families and six genera were significantly positively associated with wildfire severity. Many of these taxa contain species that are known to be critical contributors to maintaining global nitrogen cycles. NosZ and NirS nitrifying genes had significantly lower transcription rates in high-severity samples compared with control and low/moderate-severity samples. Collectively, these results suggest that high-severity wildfires significantly negatively alter the community structures and functions of nitrogen-cycling bacteria, which may have significant ramifications for ecosystem recovery in postwildfire landscapes.
Introduction
Wildfire frequency and acreage burned in the United States has increased since 1986, with more intense and frequent wildfires occurring in the western United States (Keane and Karau, 2010). The United States has experienced increases in the number of wildfires (Moritz et al., 2012), the length of the wildfire season (Westerling et al., 2006), and the affected burn area (Dennison et al., 2014). Soil bacteria are often affected by wildfires, both directly through cell death (Kara and Bolat, 2009) and indirectly owing to changes in soil characteristics postwildfire that affect metabolic rates and viability (Whitman et al., 2019). Bacteria manage nutrient cycling in soils and sediments across the globe (Kara and Bolat, 2009; Moreira et al., 2020). As a result, when the structure (i.e., species diversity and abundance) or function (i.e., genetic diversity and resilience) of soil microbiomes is changed by wildfire exposure, there can be long-lasting effects on the surrounding ecosystem. Soil bacteria can have varying responses to wildfires depending on wildfire severity, ranging from slightly diminished metabolic activity to cell death (Frangieh et al., 2020; Keeley, 2008; Robichaud et al., 2000; Sousa, 1984).
Changes in soil physiochemical characteristics from wildfires, such as pH, composition, porosity, and available nutrients, can persist for several years after initial wildfire events (Xu et al., 2012). Alterations to soil pH, nutrient bioavailability, habitat loss, and erosion can have indirect effects on bacterial biomass (Terzano et al., 2021), and increasing wildfire severity tends to directly influence these properties (Kranz and Whitman, 2019; Moya et al., 2021; Xu et al., 2012). Moderate severity wildfires can result in long-term increases in soil pH and available phosphorus and decreases in soil organic matter (SOM), moisture content, and total nitrogen (Xu et al., 2012). High-severity wildfires can cause negative effects on plant and soil properties with changes in nutrient levels (Gray & Dighton, 2009), pH, and porosity (Terzano et al., 2021), leading to the development of unfavorable environmental conditions and cell death. Nitrogen-fixing gene abundance (Nelson et al., 2021) may also be negatively impacted, indicating that severe wildfires are likely to have negative impacts on ecosystem recovery by damaging bacterial viability and metabolic activity (Pausas and Bond, 2020; Pereira et al., 2021).
Soil bacteria are responsible for cycling of essential nutrients like nitrogen (Killpack and Buchholz, 1993), sulfur (Edwards, 1998; Gu et al., 2017), and phosphorus (Mackey and Paytan, 2009). Some species of bacteria can accomplish multiple steps of the nitrogen cycle simultaneously (Kuypers et al., 2018). Thus, the reduction in one taxon can have negative effects on multiple steps in the nitrogen cycle. Bacteria are key in transforming sulfur and phosphorus into forms that plants can use, a process that is essential for ecosystem health (Edwards, 1998; Gu et al., 2017; Mittal et al., 2019; Wan et al., 2020). However, different wildfire severities are shown to have disparate effects. Low-severity wildfires can transform organic nitrogen into ammonium and nitrate, likely benefitting bacteria that utilize these nutrients, whereas higher temperatures vaporize nitrogen (Knicker, 2007). Phosphorus and sulfur are more temperature resistant, combusting at 760°C and 800°C, respectively (King et al., 2013; Knicker, 2007), so the highest losses of phosphorus and sulfur likely occur with high-severity wildfires.
Previous studies have shown that bacteria have varying responses to wildfire severity, and community recovery postwildfire is not static (Lopez et al., 2024; Stork et al., 2023). Several studies have found little significant change in bacterial distribution between burned and unburned soils following low-severity wildfires (Certini et al., 2021; Cox et al., 2022; Kranz and Whitman, 2019), whereas others have demonstrated that bacterial microbiomes experienced significant shifts throughout a 15-month postwildfire recovery period (Xu et al., 2012). Individual taxa also demonstrate unique responses; ammonifying taxa numbers increased one month postwildfire but returned to levels similar to controls after one year, whereas nitrifiers remained unchanged throughout the same time scale (Acea and Carballas, 1996). Experimental fires further demonstrate the importance of fire severity in influencing cell growth, with high-severity high-temperature fires significantly negatively impacting cell densities following SOM destruction (Bárcenas-Moreno and Bååth, 2009). Negatively impacted taxa include members of the Bacteroidetes, Acidobacteria, Chlamydiae, and Elusimicrobia phyla (e.g., Pseudomonas spp., Serratia spp., Enterobacter spp., Burkholderia spp., Klebsiella spp., and Pantoea spp.) (Adkins et al., 2020; Nelson et al., 2021).
Wildfire impacts to soil microbiomes are likely to worsen as wildfire frequency and severity increase. Significant shifts to community structures, overall biomass viability, and metabolic activity may have bottom-up effects on ecosystem homeostasis, particularly during postwildfire recovery periods. To this end, this study evaluated the bacterial community fingerprints and distribution of nitrogen-cycling taxa in soils one year after exposure to wildfire at locations of varying wildfire severity. The underlying hypothesis is that high-severity wildfires will cause the greatest reduction in nitrogen-cycling bacteria abundance and gene expression activity postwildfire.
Materials and Methods
Soil collection and wildfire severity assessment
Soil samples were collected from 11 sites affected by the Woolsey Fire in the Santa Monica Mountains National Recreation Area in California. The Woolsey Fire began on November 8, 2018, and by the time it was contained on January 4, 2019, it had burned 94,946 acres (Cal Fire Database, 2021). Soils were collected in January 2020 from areas exposed to low (0.01–0.026 δNBR), moderate (0.027–0.065 δNBR), and high (
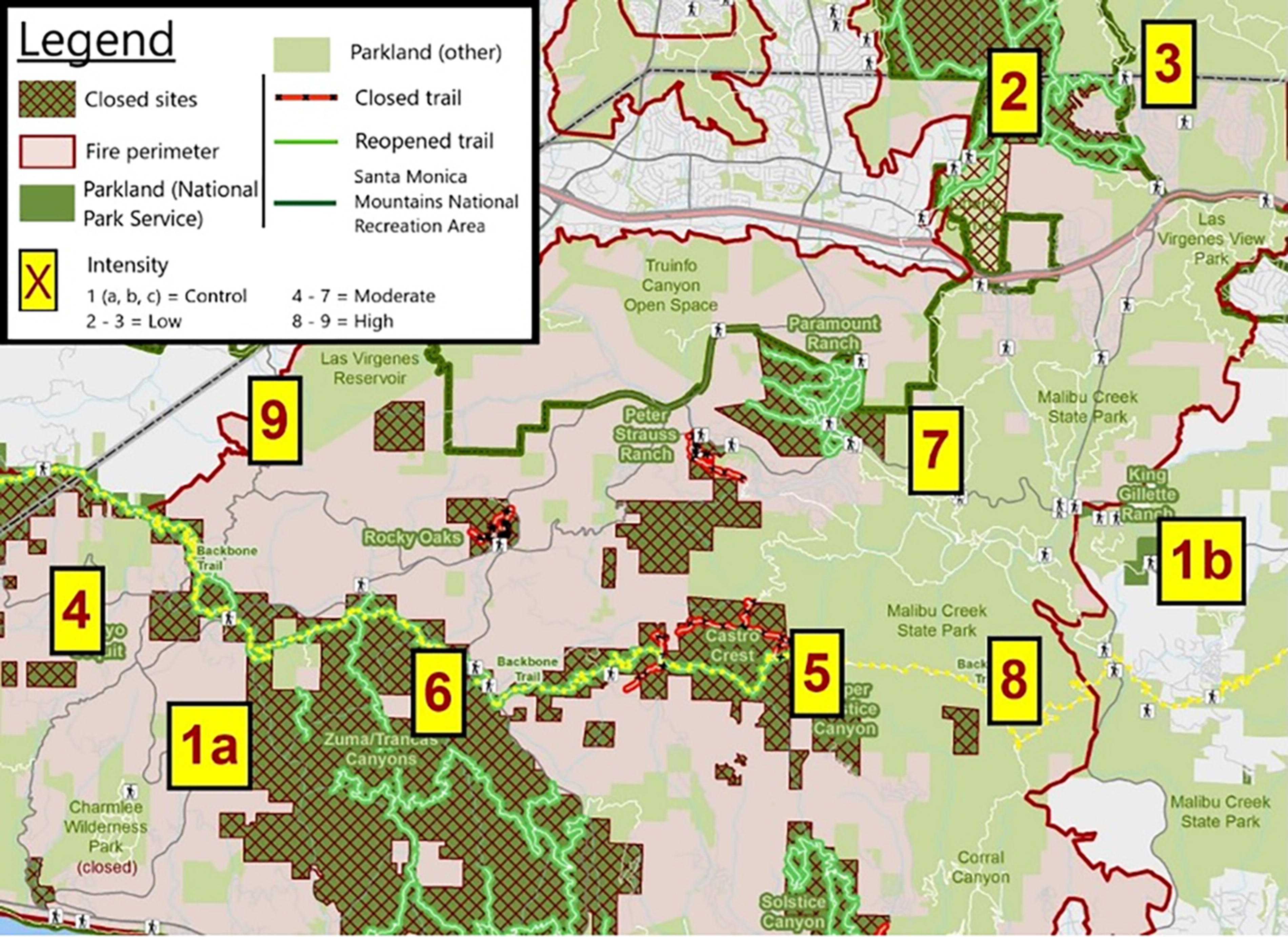
Supplementary Figure S1. Sampling locations. Labels: 1 = control sample (i.e., no fire exposure), 2–3 = low-severity sample, 4–7 = moderate-severity sample, 8–9 = high-severity sample.
Microbiome and gene expression analysis
DNA and RNA were isolated from each sample using the DNeasy PowerLyzer PowerSoil and RNeasy PowerSoil Total RNA kits, respectively, following kit manufacturer instructions (QIAGEN, Hilden, Germany). Isolated total RNA was immediately converted into cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Grand Island, NY) according to manufacturer instructions, and cDNA was purified using the DNA Clean & Concentrator-5 kit (Zymo Research, Tustin, CA). Final DNA and cDNA concentrations were determined using the Qubit 4 Fluorometer from Thermo Fisher Scientific. Purified nucleic acids were stored at −20°C for downstream analysis. For prokaryotic community structure analysis, extracted genomic DNA was used to prepare a 16S rRNA library for each sample following the protocols outlines by the 16S Metagenomic Sequencing Library Preparation guide (Illumina Inc, 2013). Prokaryotic communities were targeted using 16S rRNA primers (Gardner et al., 2020). Samples were normalized, pooled, and run on a 300 bp paired-end MiSeq platform using V3 sequencing technology at the University of Washington. After sequencing, the demultiplexed raw data was trimmed, filtered, and analyzed using QIIME 2™. Taxonomic identities were assigned using the 2013 Green Genes database. Data analysis focused on aggregated bacterial taxa with at least 10−4 relative abundance. Raw fastq files and metadata for each sample have been uploaded to NCBI under publicly available BioProject PRJNA1065882.
Because amplicon-based metagenomic sequencing offers little insight into microbiome function, we further assessed the bacterial microbiome nitrogen-cycling capacity in soil samples using quantitative polymerase chain reaction (qPCR) (i.e., g for gene abundance) and quantitative reverse transcription polymerase chain reaction (qRT-PCR) (i.e., for transcriptomic activity). Nitrogen-cycling gene abundance and expressions levels for denitrification (nirS, nosZ) and nitrification (amoA) genes were selected for qPCR and qRT-PCR. 16S quantitation was also used as a proxy for bacterial abundance. Both qPCR and qRT-PCR approaches were used to distinguish between gene abundance and gene activity, respectively. TaqMan assays were chosen for these analyses for the increased assay specificity and lower potential for overestimation relative to SYBR Green assays. Primer and probe sequences as well as assay conditions for each gene are presented in Supplementary Table S2. All assays were run in triplicate on a Bio-Rad CFX-96 Touch and included both positive and negative controls.
Statistical analysis
For Illumina Miseq data, nonparametric Spearman analysis (rS) was used to determine whether abundance was correlated with wildfire severity. A Jonckheere–Terpstra (z) nonparametric test was also conducted to further evaluate Spearman correlations results and to conduct post hoc pairwise comparisons to determine significant alterations in taxa relative abundance across severity categories (i.e., low, moderate, and high). The Jonckheere–Terpstra test has been used previously for sequencing analysis to identify statistically significant associative relationships while accounting for nonnormal distributions of taxa abundance (Cordero et al., 2021; Wijetunga et al., 2016). Tukey box and whisker plots further assessed the distributions of taxa significantly associated with wildfire severity. This approach allows for the identification of potential optimal severities (i.e., taxa that thrive in low or moderate severities) or nonlinear associations between severity and abundance. Resulting qPCR and qRT-PCR data were normalized to gene copies/g soil or gene transcripts/g soil, respectively. Significant differences in gene abundance or expression in wildfire-exposed soils were determined via ANOVA and t-test analyses. Statistical significance was assigned at p < 0.05, and significant correlations were assigned at |rS|> 0.40. Relevant comparisons were analyzed for false significance using a Benjamini−Hochberg correction analysis (q < 0.05).
Results and Discussion
Microbiome of Wildfire-Affected soils
More than 6.4 million raw reads were obtained from Illumina sequencing, with an average of 119,000 reads per sample. Approximately 4.9 million total reads remained following quality assurance steps, corresponding to 391 genera identified postaggregation. Soil bacterial communities were diverse across all Woolsey samples, with significant differences observed for communities exposed to high-severity wildfire relative to Controls (Fig. 2A–B). At the family level (Fig. 2A), control, low-severity, and moderate-severity soils were largely dominated by the same five taxa including Chthoniobacteraceae (average relative abundance: control = 5.48% ± 1.47%; low = 6.42% ± 2.30%; moderate = 7.44% ± 4.20%), Conexibacteraceae (control = 5.59% ± 0.63%; low = 4.46% ± 0.87%; moderate = 3.75% ± 1.46%), Nocardioidaceae (control= 3.77% ± 0.79%; low = 5.26% ± 0.93%; moderate = 2.95% ± 1.44%), and Rubrobacteraceae (control = 3.30% ± 0.14%; Low: 4.75% ± 1.20%; Moderate: 0.53% ± 0.41%) families. However, soils exposed to high-severity wildfire were dominated by different taxa in both top and bottom soils: Micrococcaceae (6.37% ± 5.54%), Bacillaceae (5.13% ± 1.24%), and Oxalobacteraceae (5.32% ± 4.02%). Bacterial microbiomes also tended to be less diverse in high-severity soils relative to other severities and controls, as demonstrated by the reduced number and abundance of “other” taxa outside of the top 30 families and reduced Shannon diversity indices (high = 1.23; moderate = 2.07; low = 2.20; control = 1.98; p = 0.032). Similar trends were also observed at the genus level, with delineations in community structure observed among control/low-severity soils and moderate/high-severity soils. control and Low-severity soils tended to contain high abundances of Chthoniobacter (control = 5.69% ± 1.55%: low = 2.40% ± 0.83%), Rubrobacter (control = 3.50% ± 0.15%; low = 5.08% ± 1.32%), and Conexibacter (control = 5.93% ± 0.67%; low = 4.78% ± 0.97%) genera, whereas moderate- and high-severity soils showed elevated abundances of Chthoniobacter (moderate = 7.68% ± 4.36%; high = 4.62% ± 4.09%), Arthrobacter (moderate = 4.01% ± 2.41%; High = 6.46% ± 5.66%), Bacillus (moderate = 2.56% ± 1.49%; high = 3.01% ± 2.43%) genera. Lower overall diversity was noted in high-severity samples relative to controls, though these differences were only moderately significant at the genus level (Shannon Index: high = 1.20; control = 1.72; p = 0.058). Shifts in bacterial community composition and diversity following wildfire disturbance suggest that bacteria may be very sensitive to changes in the soil ecosystem; that these changes persisted for at least a year postwildfire is further suggestive of the time required for natural soil microbiomes to recover from disturbance.
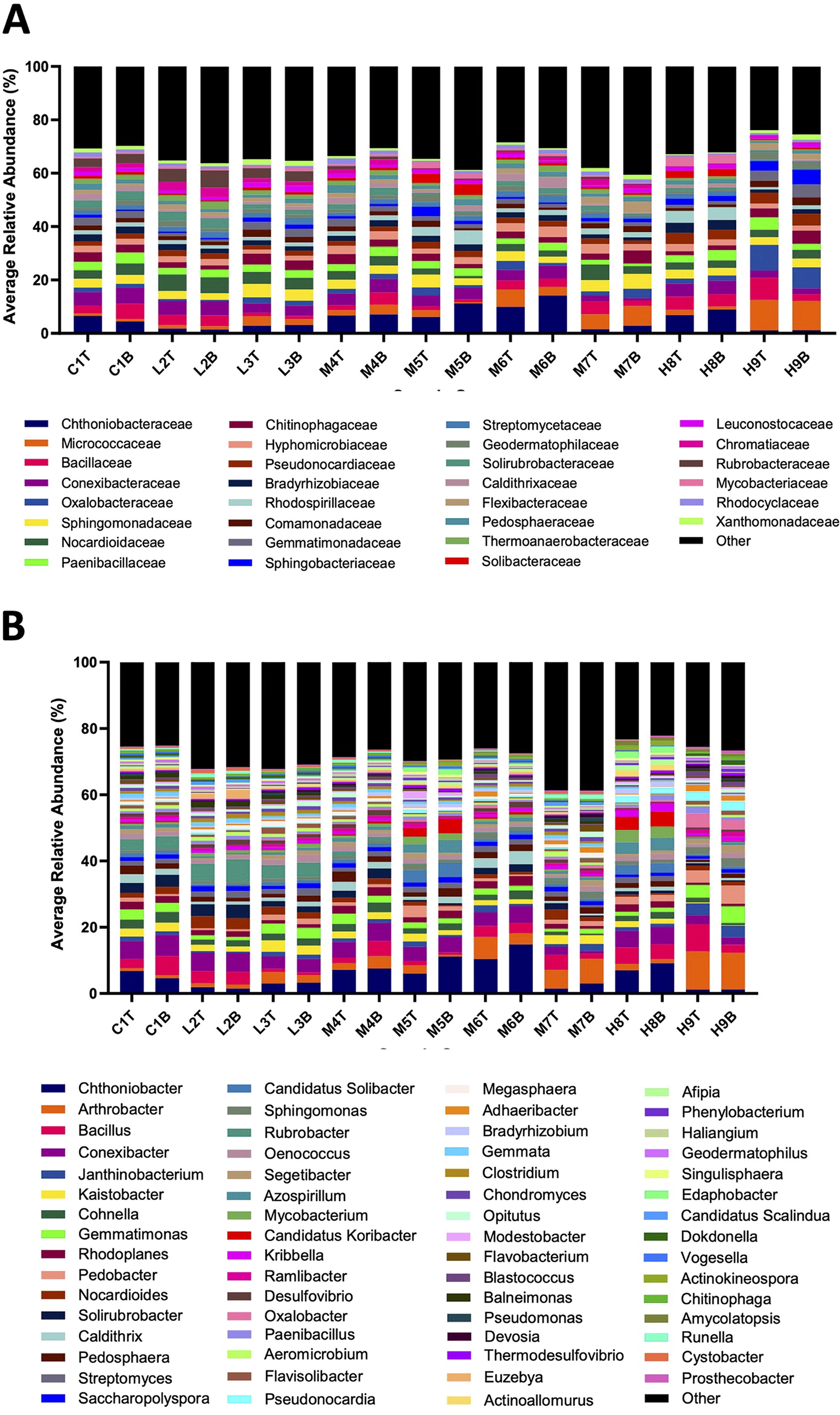
Bacterial
Of the taxa remaining post-Illumina data aggregation, 6 families and 17 genera were significantly (Spearman rs > |0.4|; p ≤ 0.05) negatively associated with wildfire severity (Table 1, Fig. 3). Although Chromaticeae abundance was only significantly reduced in soils affected by high wildfire severity relative to controls (p < 0.0001, z = 3.372), several other bacterial families were additionally significantly reduced in soils affected by moderate wildfire severity: Nocardioidaceae (p = 0.0003, z = −1.686), Rubrobacteraceae (p < 0.0001, z = −3.681), Solirubrobacteraceae (p < 0.0001, z = −2.903), Thermoanaerobacteraceae (p < 0.0001, z = −1.686), and Xanthomonadaceae (p = 0.0002, z = −1.686). These trends were magnified across genus-level taxa, where observed abundances of all 17 genera negatively associated with wildfire severity were significantly decreased in high-severity soils relative to controls, with the exception of Dokdonella, which was significantly lower only in soils affected by moderate wildfire severity. Of these 17 genera, 9 were additionally significantly decreased in moderate-severity samples, suggesting increased sensitivity to wildfire severity. One genus, Gemmata, was significantly reduced compared with controls across all three levels of wildfire severity (p < 0.0001, z = −5.141), suggesting a very low tolerance of this genus to wildfire disturbance. Conversely, the family- and genus-level taxa only significantly reduced in abundance in high-severity samples (Chromatiaceae, Candidatus Scalindua spp., Cystobacter spp., Desulfovibrio spp., Haliangium spp., Runella spp.) and suggests that these taxa may be more resilient to wildfire-induced stress. Of the six families and 17 genera negatively associated with wildfire severity, 3 of the families and 16 of the genera are associated with nitrogen cycling (Supplementary Table S3). Nitrogen-cycling taxa play a vital role in maintaining ecosystem dynamics (e.g., releasing essential nutrients for plant uptake), and their reduced abundance postfire may limit nitrogen availability in soils and slow ecosystem recovery.
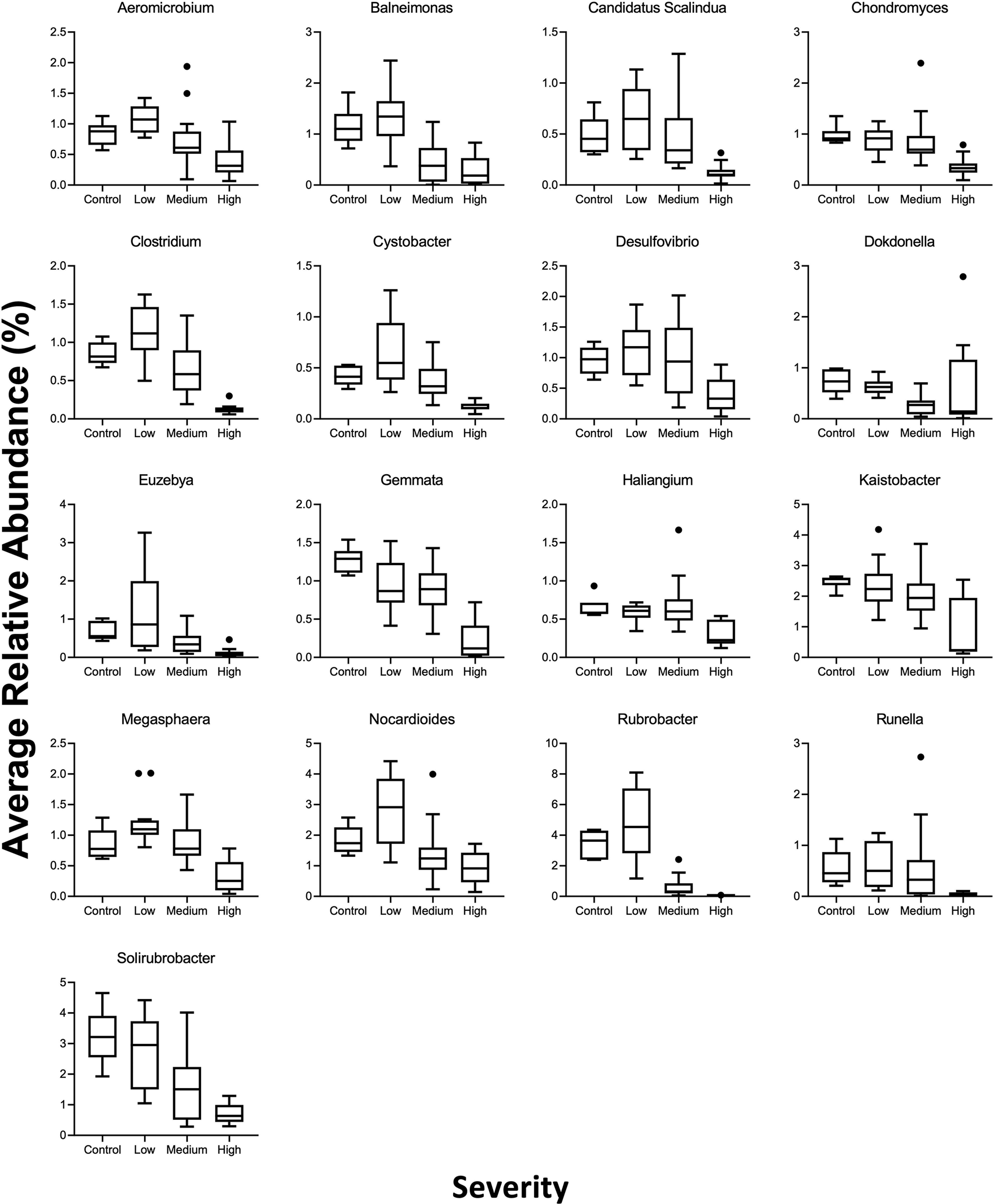
Tukey box and whisker plots of genera with significant and robust correlations (p ≤ 0.05 and |rS| > 0.4) that decrease with increasing wildfire severity.
Spearman (rs), Jonckheere–Terpstra (z), and Jonckheere–Terpstra Pairwise Comparison Tests
Control versus low severity.
Control versus moderate severity.
Control versus high severity.
Low versus moderate severity.
Low versus high severity.
Moderate versus high severity.
However, not all associations between observed taxa and wildfire severity were negative (Table 1, Supplementary Fig. S1-S2). The relative abundance of the following three families was positively associated with wildfire severity: Micrococcaceae (rS = 0.410; p = 0.0021), Pseudonocardiaceae (rS = 0.557; p < 0.0001), and Sphingobacteriaceae (rS = 0.513; p < 0.0001). The abundance of the following six genera was also positively associated with wildfire severity: Amycolatopsis (rS = 0.578; p < 0.0001), Arthrobacter (rS = 0.421; p = 0.0015), Oxalobacter (rS = 0.540; p < 0.0001), Pedobacter (rS = 0.513; p < 0.0001), Phenylobacterium (rS = 0.533; p < 0.0001), and Pseudonocardia (rS = 0.555; p < 0.0001). An increase in the abundance of these taxa may be owing to decreases in competition from taxa more sensitive to fire. For some taxa such as Chromatiaceae, abundance was only significantly increased in high-severity samples compared with controls—potentially a result of bacteria within these taxa filling the niche left by bacteria negatively impacted at high wildfire severity. The taxa that thrive postwildfire may also be better suited to the soil microenvironmental conditions determined by postfire physiochemical changes (e.g., lowered soil aeration caused by soil compaction). Four of the six genera positively associated with wildfire severity are also associated with nitrogen cycling (Supplementary Table S3). Therefore, an increase in the abundance of these genera during wildfire recovery could help to offset some of the negative effects likely to result from the reduction in other nitrogen-cycling taxa. However, nitrogen-cycling taxa negatively associated with wildfire severity outnumber those positively associated with wildfire severity four to one. Further research is needed to understand how taxa postwildfire impact the function of critical ecosystem services and postfire recovery rates.
Several taxa showed a reversal in trend, with increasing abundance relative to controls in soils affected by low wildfire severity but decreasing abundance in moderate- and high-severity samples. Taxa exhibiting this nonmonotonic trend include the Nocardioidaceae family and the genera Aeromicrobium, Clostridium, Megasphaera, and Nocardioides. Previous research supports the beneficial effects of low-severity wildfires to soil microbiomes (Moya et al., 2021), although the specific effects can be difficult to tease apart. The taxa exhibiting a trend reversal are associated with nitrogen cycling (Supplementary Table S3) and so may benefit from the nutrients transformed at lower temperatures, but then decrease in abundance when higher temperatures volatilize nitrogen (Knicker, 2007). This nonmonotonic trend may also be indicative of a decreased vulnerability of some genera to heat. For example, the genus Clostridium produces resistant spores, enabling them to survive at 100–120°C (Theodorou and Bowen, 1984). Understanding the dynamics of microbial community structure following disturbance may enable better restoration management. However, it can be difficult to parse the contributions of individual taxa in postfire ecosystem recovery.
Nitrogen-cycling gene abundance and expression
To better elucidate the functional implications of shifts in bacterial community structures within wildfire-affected soils, the current study evaluated the abundance and expression of genes that code for key enzymes of the nitrogen-cycling process. The abundance and expression of nitrogen-cycling genes in postwildfire landscapes varied significantly with exposure to increasing wildfire severity (Fig. 4). Among surveyed denitrification genes, nirS, a nitrite reductase gene, was observed most frequently within controls and wildfire-exposed soils. Distribution of nirS was highest in control top soils (avg. 4.37 × 104 copies/g) and was observed in similar numbers within the majority of top soils from low- and moderate-severity samples (range: avg. 1.87 × 103 copies/g (M4B)–7.21 × 103 copies/g (L3B), p > 0.05). However, nirS copy numbers were significantly lower in upper soils for these same sites (range: avg. 1.70 × 101 copies/g (M4T)–8.67 × 102 copies/g (C1T), p = 0.009). NirS abundance in top soils from high-severity samples also appear similar to top soils from low, moderate, and control samples. Although denitrification rates are likely to be naturally lower in oxygen-saturated upper soils, nirS abundances in low- and moderate-severity upper soil samples were significantly lower than control upper soil samples (p = 0.0072) indicating that wildfire exposure may also be negatively affecting nitrogen-cycling processes, particularly within upper soil layers. NirS copy numbers in upper soils from high-severity samples were similar to controls (p > 0.05), with the exception of H8T (p = 0.011), suggesting that relationships among nitrogen cyclers and wildfire exposure are affected by factors other than severity alone.
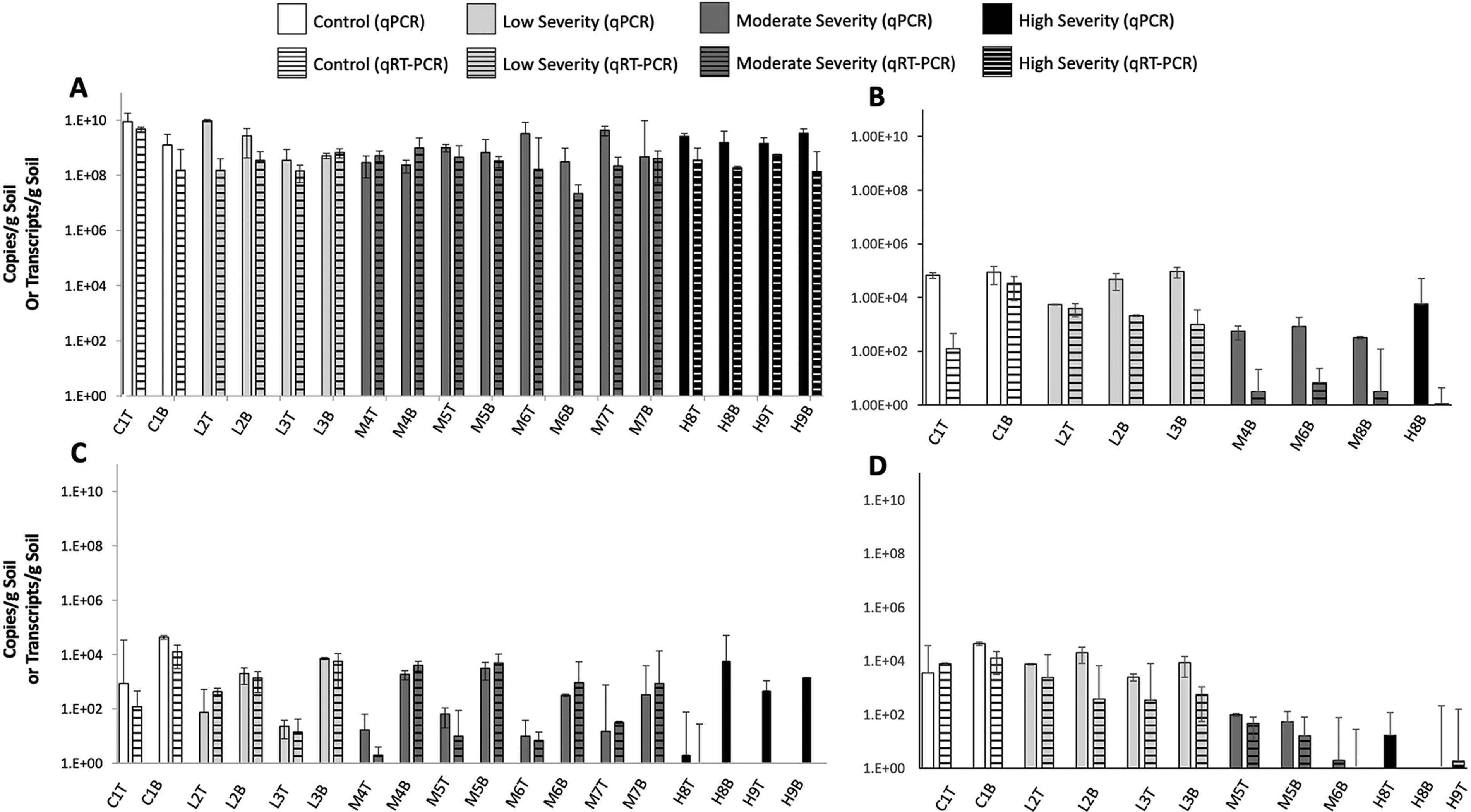
Distribution and expression of 16S and nitrogen-cycling genes in soils exposed to varying wildfire severities. Average copies or transcripts per gram of dry soil is presented for
When comparing nirS copy numbers with transcription activity (determined via qRT-PCR), different trends emerged. Within individual sampling sites (e.g., M4B), nirS transcripts/g soil were well correlated to nirS copies/g soil for both upper and lower sampling depths (p > 0.05 for all control, low, and moderate samples). However, no nirS transcripts were detected in many high-severity samples despite large observed copy numbers and average 16S transcripts >1.36 × 108 per gram of soil. This suggests that a large fraction of the observed nirS copies may reside within dead or metabolically compromised denitrifiers. This also highlights the importance of quantifying transcriptional activity, as gene abundance data alone did not indicate a significant impact to the nirS denitrification gene in high-severity soils.
NosZ, an N2O reductase gene also involved in denitrification, was impacted at both the gene copy and transcriptional level with increasing wildfire severity. The abundance and expression of this gene in moderate- and high-severity samples were consistently lower or absent relative to controls (p < 0.05; Fig. 4B). The expression of nosZ generally correlated with observed copy numbers for most control and low-severity samples except for sample C1T, which contained significantly fewer active transcripts (C1T avg. 6.77 × 104 copies/g; avg. 1.23 × 102 transcripts/g; p = 0.048). Although L2B and L3B transcripts were also lower, these differences were only moderately significant (p = 0.072 and 0.085, respectively). However, nosZ was not detected within three of the six moderate- and high-severity sampling sites. When nosZ was detected, it was only within lower depths, and expression was consistently several orders of magnitude lower than copy numbers might suggest (e.g., M4B: avg. 5.65 × 102 copies/g; avg. 3.14 × 100 transcripts/g; H8B: avg. 5.72 × 103 copies/g; avg. 1.08 × 100 transcripts/g). Both nosZ abundance (p = 0.024) and transcription activity (p = 0.003) were significantly lower in these samples relative to controls.
These trends were also observed for the amoA nitrification gene responsible for ammonia oxidation (Fig. 4D), where amoA copies and transcripts were less frequently detected in soils previously exposed to moderate and high-severity wildfire (p < 0.05). However, the amoA was more consistently detected at similar levels within both lower and upper soil depths. This may be owing to the different preferred environments for nitrifiers compared with denitrifiers—organisms that express the amoA gene are nitrifiers that tend to be more active in aerobic surface soils, whereas denitrifiers (e.g., nitrite-reducers expressing nosZ and nirS) prefer anoxic environments. These trends were not seen among 16S gene abundance and expression data, which remain consistent across sampling locations and depths (Fig. 4D). Gene copies of 16S ranged from an average of 2.35 × 108 (M4B) to 8.69 × 109 (C1T) copies/g soil and 16S transcripts ranged from an average of 2.19 × 107 (M6B) to 4.69 × 109 (C1T) transcripts/g soil. Therefore, observed differences in nitrogen-cycling gene abundance and expression within wildfire-affected soils cannot be attributed to changes in bacterial abundance.
NirS and nosZ both serve as molecular markers for denitrifying bacteria; their limited expression in higher severity wildfire-affected soils indicates that soil bacteria are likely constrained by nitrogen in these environments. A greater abundance of the nirS gene relative to the nosZ gene could indicate that these soils are still in the early stages of postfire recovery, as the nirS gene is more likely to be observed at the early stage of primary succession (Kandeler et al, 2006). NosZ denitrifiers are driven largely by the abundance of organic matter, which may be partially or completely combusted by wildfire (Kandeler et al, 2006). The abundance and expression of the amoA gene, a marker for ammonia oxidizers, were similarly affected by higher severity fire, further indicating the limited availability of reactive nitrogen forms in the postfire environment. Taken together, gene abundance and expression data in the current study indicate that high-severity wildfires negatively impact the functions of nitrogen-cycling bacteria at least one-year postfire.
Conclusions
Wildfire frequency and severity has increased in recent years and additional research is needed to understand the holistic impacts of wildfires on environmental health and sustainability. Although large-scale effects of wildfires are well studied (e.g., erosion, property destruction, and loss of animal habitats), the more subtle effects of wildfires have yet to be fully evaluated. Owing to the importance that bacteria play in maintaining healthy ecosystems, negative impacts on soil communities from wildfires have the potential to pose a serious threat to postwildfire recovery efforts for years after a wildfire. The abundance of 6 families and 17 genera was negatively associated with wildfire severity, and the abundance of 3 families and 6 genera was positively associated with wildfire severity. Many genera that decreased with wildfire severity are known to be critical contributors to maintaining global nutrient cycles. These data suggest that high wildfire severity is detrimental to most nitrogen-cycling taxa, potentially owing to diminished nutrients or cell death from wildfire heat. Thus, the present research addressed the goal of identifying wildfire-mediated changes in the populations of families and genera in the soil.
Although the data presented here offer novel insights into the dynamics between soil bacteria and wildfires, there are several study limitations that affect their potential impact. Satellite measurements gauged severity but did not include capturing heat transfer or soil temperature data. Fire characteristics (e.g., surface vs treetop burning) were not recorded, thus low-severity surface fires could have caused greater soil temperatures than high-severity fires. Another limitation of the study was that control samples were not collected before the fire. Before sampling, bacteria could have been transported between burned and unburned locations through runoff, air transport, and foot traffic, potentially affecting the sequencing results. This study also focused exclusively on soil bacteria, leaving the responses of microbial eukaryotes unexplored. However, study findings will increase the knowledge of bacteria families and genera that are affected by wildfires, enabling a more holistic understanding of the impacts of wildfire on ecosystem recovery. Although the findings from this research expand upon the current knowledge of impacted taxa from wildfires, further research is needed. Samples were collected one year postwildfire during the bacterial community recovery period. Future research should investigate long-term changes to microbial communities across a range of wildfire severities coupled with severity data to further categorize microbial community response to wildfire impacts to better determine ecosystem recovery needs and timelines. Future research should also assess the potential shifts in the metatranscriptome and include the responses of microbial eukaryotes (e.g., fungi) to wildfire as well as the contribution of microbial eukaryotes to the trends observed here.
Footnotes
Authors’ Contributions
T.P.: Formal analysis; investigation; data curation; writing—original draft (lead); writing—review and editing; and visualization. L.M.: Writing—original draft (supporting) and writing—review and editing. J.E.: Investigation and data curation. C.G.: Conceptualization; methodology; investigation; writing—original draft (supporting); writing—review and editing; visualization; supervision; and project administration.
Author Disclosure Statement
The authors declare no conflicts of interest for this work. The authors also confirm that the article has not been published elsewhere and is not under consideration for publication elsewhere. All research procedures were conducted in accordance with the ethical standards of Washington State University and the University of Texas at Austin.
Funding Information
The authors have no funding information to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.