
Abstract
Widespread use of antibiotics has led to an increase in the virulence factors (VFs) and antibiotic resistance genes (ARGs) within soil microbial communities. However, the impact of agricultural management, plant type and soil compartment on the distribution of VFs and ARGs in both organic systems and conventional systems remains unclear. In this study, we conducted a metagenomic analysis to investigate the distribution. Nonmetric multidimensional scaling analysis revealed that soil samples exhibited clustering primarily based on agricultural management system for both VFs (stressVFDB = 0.071) and ARGs (stressARDB = 0.117, stressCARD = 0.069). We observed significantly higher levels of VFs and ARGs in soils from conventional systems compared with organic systems (p < 0.05). Furthermore, combined analysis of carbohydrate-active enzymes (CAZymes) and KEGG revealed that six classes comprising 445 families of CAZymes were significantly more abundant in conventional system soils than in organic system soils. Notably, the abundance of carbamyl phosphate synthase (ammonia) was relatively higher in organic system samples (53.33%) compared with conventional system samples (48.8%). Our comprehensive analysis suggests that different agricultural management practices lead to alterations in soil microbial communities. Specifically, organic management practices were found to effectively mitigate the pathogenic potential and drug resistance of soil microbial communities. In conclusion, our findings highlight the substantial influence of agricultural management on soil microbial communities, with corresponding clustering patterns observed in VFs and ARGs. Organic farming practices contribute to the promotion of healthier soil microsystems and the sustainable development of agriculture compared with conventional systems.
Introduction
In agroecosystems, the adoption of various agricultural management practices, such as fertilization, tillage, and pest control measures, plays a pivotal role in shaping the diversity and composition of soil microbiota (Zhou and Fong, 2021). Conversely, the presence of pathogenic microorganisms, including bacteria, viruses, fungi, and protozoa, poses a constant threat to agroecosystems by causing diseases through invasion, colonization, and subsequent damage. Widespread use of antibiotics has exacerbated the evolution of highly resilient pathogens, posing a significant risk to human health due to the coselection of virulence factors (VFs) and antibiotic resistance genes (ARGs) (Pan et al., 2020). Meanwhile, antibiotic residues are transferred to aquatic ecosystems, and municipal waste discharge and its use in crop irrigation pose a significant problem, leading to soil pollution (F. Z. Vilca et al. 2024). VFs encompass various molecular components that enhance a microorganism’s capability to establish itself on or within a host and heighten its potential to cause disease. These components include bacterial toxins, cell surface proteins facilitating bacterial attachment, protective cell surface carbohydrates and proteins, as well as hydrolytic enzymes contributing to bacterial pathogenicity (Chellapandi and Prisilla, 2018). Antibiotic-resistant bacteria in soil pose serious health risks (Hashmi et al., 2017; Zhu et al., 2022). ARGs from antibiotic-treated animals’ manure can enter the food chain via agricultural soils (Gillings, 2013). Carbohydrate-active enzymes (CAZymes) constitute a group of enzymes and related active substances capable of degrading, modifying, and synthesizing glycosidic bonds. CAZymes degrade glycosidic bonds, pivotal for glycogenomics and understanding microbial biomass conversion potential (Andrade et al., 2017; Cantarel et al., 2009; Wardman et al., 2022). The relationship between resistance genes that enter the food chain through soil transmission and CAZymes is an important area of study in microbial ecology and public health. Resistance genes can be transferred to soil microorganisms through various means, including agricultural practices and environmental contamination. CAZymes play a crucial role in the degradation and utilization of complex carbohydrates, impacting microbial community composition and function in the soil (Xiao et al., 2023).
In recent years, VFs, ARGs, and CAZymes have been found in various environments, including wastewater, coastal beaches, aquaculture, as well as glacial soil, permafrost, and sediments (Fresia et al., 2019; Su et al., 2020; Tang et al., 2021; Zhang et al., 2018). In aquatic environments, xenobiotic pollution affects ARG and VF transcription (Zhang et al., 2022), while in terrestrial settings, ARG abundance rises with prolonged sewage sludge and pig manure application (Chen et al., 2016a, 2016b; Peng et al., 2017). Pig manure notably increases the presence of integrase gene I1, associated with ARGs (Peng et al., 2017).
In agricultural systems, the ideal scenario involves sustainability, human health enhancement, environmental benefits, and meeting global food demands (Singh et al., 2011). Organic agriculture prioritizes factors beyond economic profit, whereas conventional agriculture relies on chemical inputs for higher yields (Shennan et al., 2017). Comparing organic and conventional farming systems provides insights into productivity and environmental impact. Real-world agroecosystem monitoring offers a viable alternative, considering diverse factors such as location, soil type, and cropping systems (Shennan et al., 2017). Our previous study explored microbial diversity in conventional and organic farming soils, finding higher diversity in organic systems, indicating environmental benefits (Zhou and Fong, 2021). We also identified elevated levels of human pathogen-related Vibrio cholera and resistance to antimicrobial peptides in conventional soils. However, disparities in VFs and ARGs between conventional and organic systems need further investigation.
Building on our prior research, we are conducting a thorough investigation into how agricultural practices (organic vs. conventional), plant types (cabbage vs. lettuce), and soil areas (rhizosphere vs. bulk) impact the presence of ARGs and VFs in a real agroecosystem. Our hypothesis suggests that (a) agricultural practices have the most significant impact and (b) differences in ARGs and VFs reflect adaptive responses from microbial communities to these factors. To test these ideas, we are performing a metagenomic analysis of soil microbiota in two soil areas (bulk and rhizosphere) and two plant types (cabbage and lettuce) across two different agricultural management systems (conventional and organic).
Material and Methods
Study site and experimental design
The samples used in this study match those in Zhou and Fong (2021), with key details summarized here, while the original publication contains more specifics. The research site is in Tsang Uk Tsuen, Shek Kong, Yuen Long District, New Territories, Hong Kong (22.427920N and 114.092972E). This site comprises 21 privately-owned agricultural land lots. We focused on two neighboring lots (<60 m apart) under different ownership—one practiced organic agriculture (>10 years) and the other conventional methods (>40 years). We selected these lots because (1) we had permission, (2) they cultivated the same crops, (3) their farming practices were well-documented and reliable, and (4) their close proximity minimized differences in unrelated factors.
Each land parcel is subdivided into smaller sections (approximately 20 × 1 m) dedicated to the cultivation of different crops. As these are nonexperimental plots, historical information regarding previous crops grown prior to the study period is unavailable. The organic and conventional land parcels varied in their methods of fertilization, soil preparation, and plant protection. The two crops selected were cabbage (Brassica oleracea var. capitata) and Indian lettuce (Lactuca indica).
We sampled soils in January 2020 from two soil compartments (rhizosphere and bulk soil) just before the harvest of both crops. Rhizosphere soil (soil adhering to the root crowns) was collected by removing a randomly selected plant and associated root crown (depth 5–20 cm), lightly shaking to remove soil not associated to the roots, then collecting soil attached to roots. Bulk soil was collected adjacent to the plant (5–20 cm) using a soil sampling tube (LaMotte, #1055). A total of 24 soil samples were collected (3 individuals × 2 plant types × 2 soil compartments × 2 agricultural management systems). Soil samples are named as plant (cabbage [C] or lettuce [L]), soil compartment (bulk [B] or rhizosphere [R]), and agricultural management system (conventional [C] or organic [O]). For instance, the “CBC” sample is from cabbage, bulk soil, conventional, while the “LRO” sample is from lettuce, rhizosphere soil, organic.
DNA extraction and metagenomic analysis
DNA extraction was performed using 0.5 g of soil with the FastDNA Spin Kit (MP Biomedicals, OH, USA). Metagenomic DNA was detected using 1% agarose gel electrophoresis and DNA libraries were subsequently constructed from this purified DNA. High-throughput sequencing operations and quality control were conducted at Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). Sequencing of samples yielded an average sequence read depth of 7.5 × 107 per sample (min = 6.7 × 107; max = 8.9 × 107).
Raw metagenomic and metatranscriptomic datasets were deposited in the NCBI Sequence Read Archive database (accession number PRJNA852672). General sequencing statistics for all samples and mean sequence quality distribution were measured with FastQC (v.0.11.6). Residual primer and adapter content were removed using the Joint Genome Institute Bestus Bioinformatics Decontamination Using Kmers (BBDuk) tool (v.38.07). Reads were trimmed at both ends until the mean Phred quality score is at least 17 for at least 80% of the read length for all reads of all sample libraries. After trimming, the average sequencing read length across libraries was 150 bp.
The filtered, high-quality sequences were assembled into predicted gene sequences. These predicted gene sequences were aligned to the nr database (NCBI) to obtain species annotation information, nonredundant gene sequences to the database, and annotations based on functional domains with functional similarity between sequences. Subsequently, we conducted follow-up functional analyses, environmental factor association analysis, and correlation and model prediction analysis.
VF and ARG analysis
We used the Virulence Factor Database for analyzing VFs, a specialized resource for pathogenic bacteria study. It provides species information, descriptions of characteristics, virulence gene functions, and detailed explanations of pathogenic mechanisms.
An ARG reference database was constructed by integrating data from two commonly used databases: the Antibiotic Resistance Genes Database (ARDB) and the Comprehensive Antibiotic Resistance Database (CARD) (Yang et al., 2016). Information extracted from these databases includes the relative abundance of each ARG, species attribution, and resistance mechanisms. Initially, gene catalog data were compared with the CARD database (blastp, evalue ≤ 1e-5) using Diamond software (Version 0.9.9) (Hua et al., 2015, Yang et al., 2013). Subsequently, the comparison results were filtered based on criteria of similarity ≥90% and sequence length ≥75 bp (Bing et al., 2016). The relative abundance values of each ARG were then calculated according to the comparison results of each gene.
Contig sequences assembled were compared with the CAZymes and KEGG database using DIAMOND (Vincent et al., 2014) to analyze microbial carbohydrate active enzyme functional gene composition. Abundance analysis of CAZymes in the CAZy annotation results was done in R.
Statistical analyses
Significant differences in VFs, ARGs, and CAZymes among samples were determined using one-way analysis of variance (ANOVA). Paired comparisons of treatment means were conducted using Tukey’s procedure at a significance level of p < 0.05. Statistical analyses were performed using SPSS BASE ver. 19.1 (SPSS, Chicago, IL, USA) (Wang et al., 2020) and the results were visualized using Origin 9.0.
Results
VF analyses
We identified 238 VFs from 24 soil samples, with 29 exceeding 1% abundance (Supplementary Table S1). Comparing conventional and organic soils, significant differences emerged. In conventional soils, five defensive VFs had notably higher levels, including three antiphagocytosis (Fig. 1A–C), one cellular (Fig. 1D), and one metabolism VF (Fig. 1E). In addition, conventional soils exhibited elevated levels of four offensive VFs compared with organic soils, including two invasion (Fig. 1F–G), one adherence (Fig. 1H), one toxin (Fig. 1I), and one regulator of virulence-associated gene (PhoP, Fig. 1J).

Virulence factors statistically higher in soils from conventional management system than organic management system (cabbage [C] or lettuce [L], bulk [B] or rhizosphere [R], conventional [C] or organic [O]).
In the analysis of VFs, hierarchical clustering showed distinct sample grouping. Initially, samples were clustered by agricultural management system (organic vs. conventional) and then by plant type (cabbage vs. lettuce) (Fig. 2A). Notably, VFs did not cluster by soil compartment (rhizosphere vs. bulk). Nonmetric multidimensional scaling (NMDS) analysis supported these findings, showing robust clustering by agricultural management and plant type. NMDS1 separated samples by management system (green for conventional, blue for organic), while NMDS2 further differentiated samples by plant type (purple for lettuce, red for cabbage). The first two principal coordinates explained 66.02% (NMDS1) and 12.54% (NMDS2) of the variation, with a stress value of 0.071 (Fig. 2B).
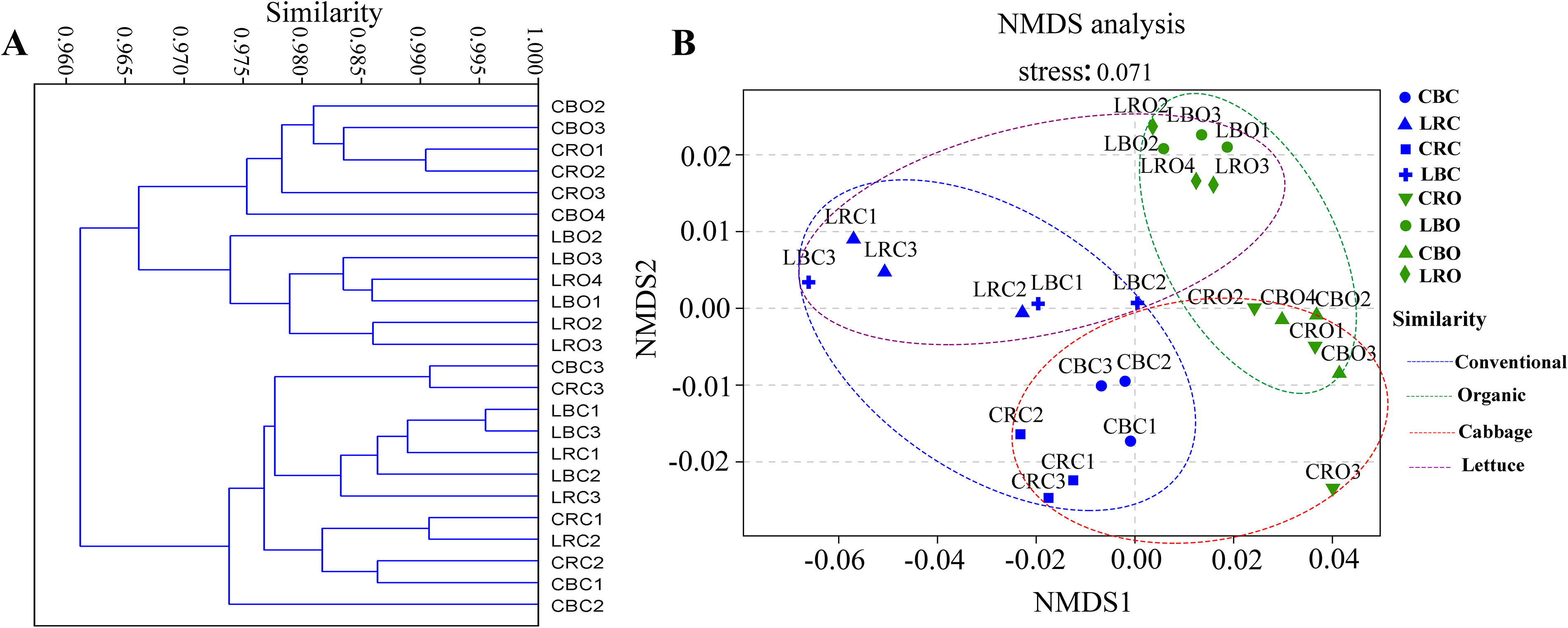
Beta diversity analyses of virulence factors.
ARG analysis
In our ARDB analysis, we found seven types of ARGs with a total of 286 subtypes across all samples. Predominant ARGs at the class level included undecaprenyl pyrophosphate phosphatase (88.39%), sulfonamide-resistant dihydropteroate synthase (2.09%), aminoglycoside O-nucleotidylyltransferase (1.49%), and resistance-nodulation-cell division transporter system (0.97%). ARG abundance was significantly higher in conventional soils compared to organic soils (p < 0.05). For instance, undecaprenyl pyrophosphate phosphatase was notably more abundant in conventional soils than in organic soils. In addition, six genes showed significantly higher expression in conventional soils for sulfonamide, tetracenomycin_c, tigecycline, streptomycin, glycylcycline, and acriflavin (see Fig. 3A–F).
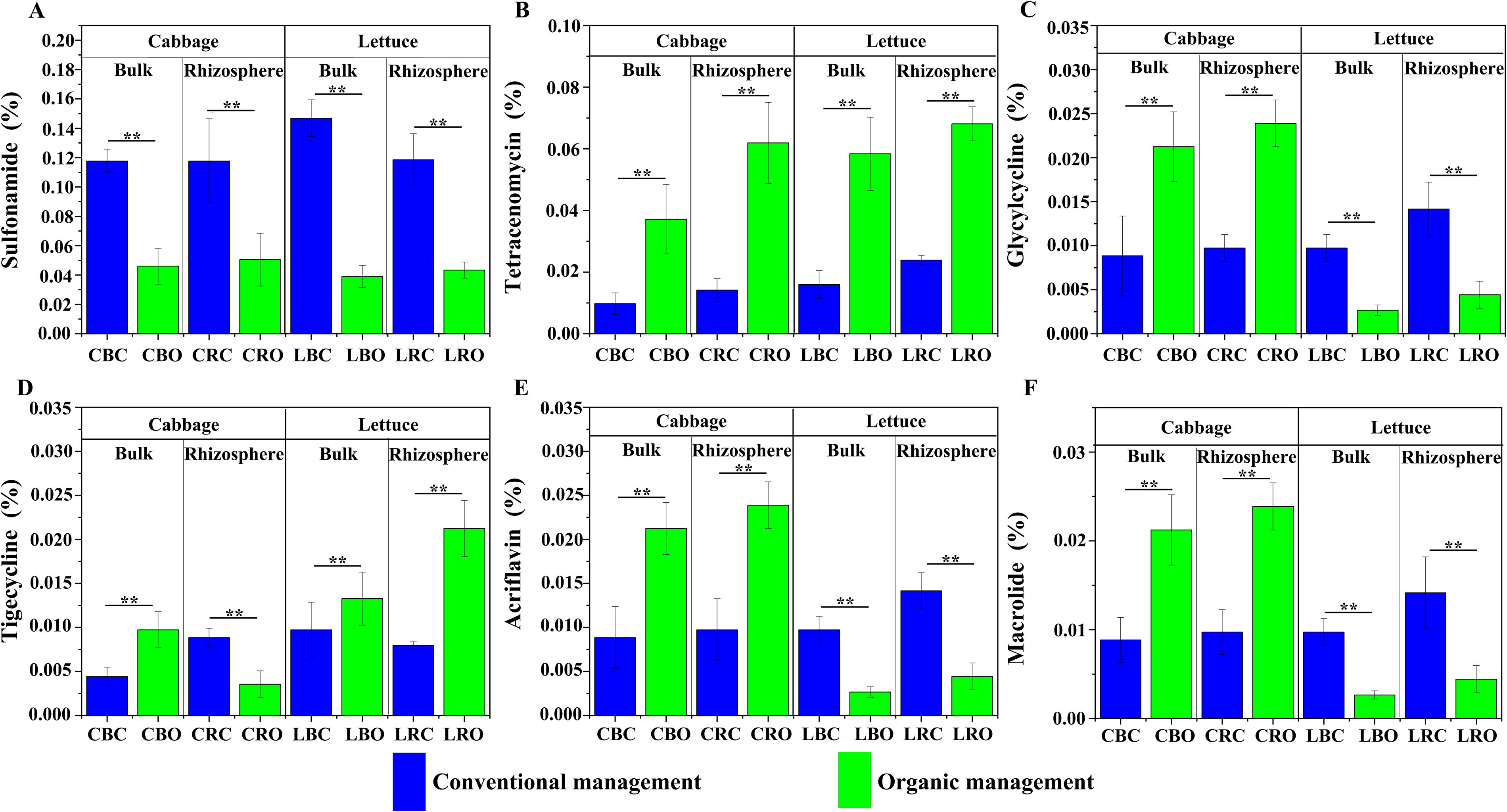
Antibiotic resistance genes (from the Antibiotic Resistant Database [ARDB]) statistically different between organic and conventional management samples (cabbage [C] or lettuce [L], bulk [B] or rhizosphere [R], conventional [C] or organic [O]).
In our analysis of the CARD, we identified 235 ARGs with significant differences across the 24 soil samples. The top two prevalent ARGs were Staphylococcus aureus rpoC, known for daptomycin resistance, and Escherichia coli rpoB mutants, linked to rifampicin resistance (refer to Supplementary Table S3). Gene abundance consistently favored higher levels in conventional system soils compared with organic system soils for genes related to antibiotic resistance, sensitivity, and targets (see Fig. 4A–C).

Antibiotic resistance genes (from the Comprehensive Antibiotic Resistance Database [CARD]) statistically different between organic and conventional management samples (cabbage [C] or lettuce [L], bulk [B] or rhizosphere [R], conventional [C] or organic [O]).
In our beta diversity analysis of ARGs, results showed strong consistency between the ARDB (Fig. 5A) and CARD analyses (Fig. 5B). Soil samples mainly clustered by agricultural management practices (organic vs. conventional), with additional clustering based on plant type (cabbage vs. lettuce).
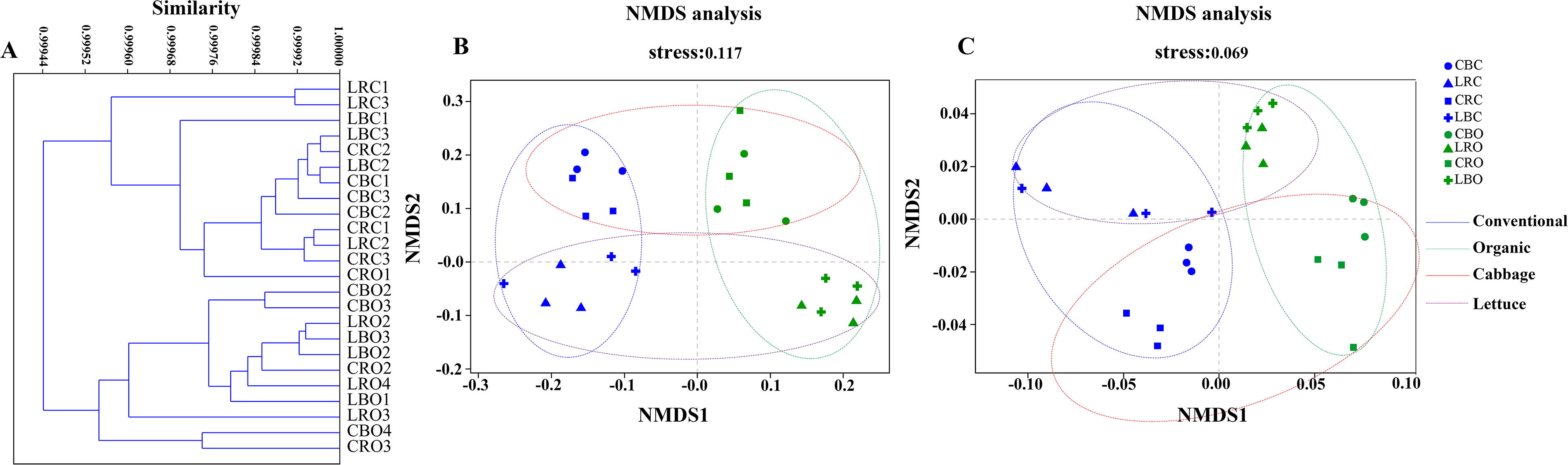
Beta diversity analyses of antibiotic resistance genes.
CAZymes
For CAZymes, we identified six classes comprising a total of 445 families of CAZymes across all samples (Supplementary Table S4). Notably, the relative abundance of these six classes starkly favored conventional system samples over organic system samples: glycoside hydrolases, glycosyl transferases, carbohydrate esterases, auxiliary activities, polysaccharide lyases, and carbohydrate-binding modules (see Fig. 6A–F).

CAZymes statistically different between organic and conventional management samples (cabbage [C] or lettuce [L], bulk [B] or rhizosphere [R], conventional [C] or organic [O]).
KEGG
In our KEGG analysis, we found 54 orthologous functional genes across all samples. While no significant differences were observed among the samples, we noted noteworthy disparities in the relative abundance of five enzymes between conventional and organic system samples. Notably, the most prevalent KEGG functional gene was carbamoyl-phosphate synthase (ammonia) [EC:6.3.4.16], with a relatively higher abundance in organic system samples (53.33%) compared with conventional system samples (48.8%, see Supplementary Table S5 and Supplementary Fig. S1A).
Discussion
VF abundance in organic and conventional systems
Microbial VFs are diverse molecules produced by pathogenic microorganisms, aiding in evading host defenses and causing disease (Leitao, 2020). The virulence regulatory network controls primary metabolism to enhance robustness upon infection (Peyraud et al., 2018). Proteins involved in antiphagocytosis, adherence, and biofilm formation are crucial VFs during bacterial infections (Carkaci et al., 2017). Yang et al. (2022) noted a negative correlation between soil electrical conductivity and the relative abundances of antiphagocytosis, secretion systems, and toxins. Virulence genes (encoding VFs) serve as indicators of bacterial pathogenic potential and often coexist with ARGs in a strong positive association (Bing et al., 2016). Our findings showed significantly higher abundances of VFs and ARGs in conventional system soils compared to organic ones, consistent with Bing et al. (2016) (Supplementary Table S2 and Supplementary Table S3). VFs are mainly influenced by nutrient variables in sediment (Lu et al., 2021). In our study, the conventional agricultural system receives synthetic and organic fertilizers, potentially increasing the abundance of genes related to ARGs and virulence due to fertilization-induced nutrient pollution (Chen et al., 2019).
Soborg et al. (2013) confirmed that occurrence of virulence genes in the environment was generally not the result of the presence of clinically relevant bacteria, indicating an environmental origin of the virulence genes. Their results also indicated that about half of the variation in the distribution of the virulence traits could be explained by differences in the pH. Specifically, higher pH values were associated with lower distributions of virulence traits. Similarly, we observed a parallel trend in our study based on pH, where higher pH levels in organic systems corresponded to lower distributions of virulence traits.
ARG abundance in organic and conventional systems
Our study found a significantly higher abundance of ARGs in conventional system soils compared with organic ones (p < 0.05) (Fig. 4B). While some studies suggest that composting increases both ARG diversity and abundance (Cheng et al., 2019; Qian et al., 2016; Sharma et al., 2009), Xie et al. (2016) observed a decrease in ARGs, from 213 targeted genes, following traditional industrial-scale composting of animal manure. In addition, soil characteristics such as texture, type, pH, moisture content, as well as the presence of antibiotics and heavy metals, indirectly influence ARG richness by affecting bacterial community succession and mobile genetic element abundance (Liu et al., 2021; Mahapatra et al., 2022), shedding light on our findings.
Fahrenfeld et al. (2014) noted a decline in some ARGs to environmental background levels within 2 months after manure application in real field conditions. Similarly, He et al. (2016) reported elevated ARG abundance and diversity in soils of vegetable fields fertilized with swine wastewater for over 24 years. Considering our organic soil samples are approximately 10 years old and the experimental conditions are stable, these factors likely influenced our observations.
In addition, we found a significantly higher sulfonamide abundance in conventional system samples compared with organic ones (p < 0.01) (see Fig. 3A). Sulfonamide, widely utilized in animal breeding due to its broad antibacterial spectrum (Jeong et al., 2010) can reach concentrations as high as 20 mg·kg−1 in animal manure (Zhu et al., 2019), potentially leading to reduced abundance and diversity of soil microorganisms in conventional systems compared with organic systems. This difference may stem from various synthetic fertilizers, particularly untreated cow manure, used in conventional systems. Direct manure application to agricultural land for nutrient recycling purposes can introduce pathogens, antibiotic-resistant bacteria, or genes into the environment (Do et al., 2022). Tran et al. (2021) confirmed that raw cow manure samples amplified some antibiotic resistance-associated gene targets. Furthermore, we found that genes related to antibiotic sensitivity and targets were significantly more abundant in conventional soil than in organic soil (see Fig. 4A, C).
Distribution characteristics of VFs and ARGs in organic and conventional systems
For both VFs or ARGs, the beta diversity analyses revealed sample separation based on agricultural management systems (organic/conventional) and plant types (cabbage/lettuce) but not soil compartments (rhizosphere/bulk) (Fig. 5). These results indicate that different agricultural management systems affect the distribution characteristics of VFs and ARGs. Previous studies have highlighted the predominant influence of environmental factors and microbial communities on the distribution of VFs and ARGs (Chen et al., 2020, Cao et al., 2020). Given that microorganisms serve as carriers of ARGs and VFs, variations in microbial community composition are pivotal in determining the profiles of ARGs and VFs (Zou et al., 2023).
Abundance of CAZymes in organic and conventional systems
CAZymes indicate the metabolic potential within a microbial community for converting biomass (Andrade et al., 2017). These enzymes, produced by various organisms, have significant environmental and biotechnological importance (André et al., 2014). Plant roots release diverse compounds, including ethylene, sugars, vitamins, amino acids, organic acids, polysaccharides, and enzymes (Garbeva et al., 2004). Changes in CAZyme abundance may result from shifts in microbial composition, with higher CAZyme levels observed in samples with consistently lower respiration rates, suggesting a potential connection between metabolic expression and long-term community composition changes (Albright et al., 2018). Our previous work has also shown differences in microbial communities between conventional and organic systems (Zhou and Fong, 2021).
Abundance of KEGG in organic and conventional systems
Our investigation revealed significant differences in the abundance of glutamine synthetase, glutamine dehydrogenase, NADH-glutamate dehydrogenase, carbamate kinase, and carbamoyl-phosphate synthase between conventional and organic systems. Here are their functions: (1) Glutamine synthetase: Crucial for nitrogen metabolism, assimilating ammonium into amino acids (Bernard and Habash, 2009). (2) NADH-glutamate dehydrogenase (GDH): A versatile enzyme with reversible activity, contributing to various metabolic processes (Marchi et al., 2021). Feng et al. (2014) showed its strategic position in the nitrogen metabolic signaling pathway, with expression potentially upregulated in response to nitrogen stimulation. (3) Carbamate kinases: Facilitate the conversion of carbamate to carbamoyl phosphate, a precursor for other compounds (Hennessy et al., 2018).(4) Carbamoyl-phosphate synthase: Initiates nitrogen disposal by producing carbamoyl phosphate from ammonia and bicarbonate in the mitochondria (Kim et al., 2017). McGivan et al. (1976) described conditions where it becomes rate-limiting, particularly in the presence of NH4Cl, ornithine, and excess ATP. Our findings suggest that different agricultural management methods can influence the key enzyme systems of soil microbial flora, further validating differences in microbial community composition between conventional and organic systems.
Conclusions
In our study, we found that agricultural management practices, whether organic or conventional, had the most significant impact on the distribution patterns of VFs and ARGs, with plant type being a secondary factor. Our KEGG functional analysis highlighted notable differences between conventional and organic systems. Interestingly, we observed a lower abundance of CAZymes in the organic system, potentially indicating reduced production efficiency. However, this was offset by significantly lower levels of VFs and ARGs in the organic system compared with the conventional one. This suggests that VFs and ARGs adapt to different experimental conditions, with beneficial soil microorganisms playing a larger role in organic systems. This aligns well with the goals of sustainable agriculture and helps mitigate public health risks. In addition, the organic management model offers other benefits, such as reducing the proliferation of pathogenic microorganisms from chemical overuse and manure application, improving crop nutrition, and lowering production costs. Thus, the organic model appears as a preferable approach in agricultural production.
Footnotes
Data Availability Statement
The data presented in this study are available in the article. The sequences obtained have been submitted to the NCBI SRA database under the accession number PRJNA852672.
Authors’ Contributions
J.J.F.: review and editing (equal). X.J.Z.: Conceptualization (lead); writing—original draft (lead); formal analysis (lead); writing—review and editing (equal). Y.W.: Software (lead); writing—review and editing (equal). J.Z.: Methodology (lead); writing—review and editing (equal). Y.K.: Conceptualization (supporting); Writing— original draft (supporting); Writing—review and editing (equal).
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This study was supported by the National Natural Science Foundation of China (No. 32170530), Lingnan University, Hong Kong Special Administrative Region, China under the Faculty Research Grant Scheme (Project No.: 102173), and the Croucher Chinese Visitorship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.