
Abstract
Background and Purpose:
Stretch-induced cyclooxygenase-2 (COX-2) expression occurs in urothelial cells during urinary tract obstruction (UTO). This increases COX-2–dependent prostanoid synthesis in stretched urothelial cells. These prostanoids then act on afferent neurons and smooth muscle cells in the ureter to amplify nociceptive and contractile responses, respectively. We previously used a unilateral ureteral obstruction (UUO) mouse model and a primary human urothelial cell (HUC) stretch model to describe ureteral COX-2 expression during UTO. The current study was performed to determine whether phosphatidylinositol 3-kinase (PI3K)-dependent signaling pathways are necessary for stretch-induced COX-2 expression in urothelial cells.
Materials and Methods:
Adult male CD-1 mice were treated with 25% dimethyl sulfoxide/phosphate buffered saline or PI3K inhibitor LY294002 (3 mg/kg, 30 mg/kg) for 1 hour before performing UUO for up to 4 hours. Obstructed and contralateral mouse ureters were analyzed via immunohistochemistry or Western blotting to assess in vivo stretch-induced COX-2 expression. In addition, HUCs were cyclically stretched (5%–20% displacement, 12 cycles/min) on collagen I-coated stretch plates and assessed for COX-2 expression via Western blotting.
Results:
Histologic analyses of obstructed ureters show that urothelial cells stretch in response to external obstruction, COX-2 expression increases in the stretched urothelial cells, and no infiltrating immune cells were present under the conditions of the study. PI3K inhibitor LY294002 (30 mg/kg) attenuated in vivo stretch-induced COX-2 expression. LY294002 or RNA-interference also attenuated (HUC) stretch-induced COX-2 expression in vitro. Furthermore, the results also show that LY294002 inhibits stretch-induced protein kinase C (PKCζ) activation previously identified upstream of stretch-induced COX-2 expression in HUCs.
Conclusions:
The results indicate that PI3K is a mediator of stretch-induced COX-2 expression in urothelial cells. Identifying molecules that couple urothelial cell stretch to COX-2 expression may provide targets of drug action for effective therapeutics for UTO.
Introduction
Cyclooxygenase (COX) catalyzes the rate-limiting step of prostanoid synthesis in urinary tissues and exists in two isoforms as COX-1 and COX-2. 3 Conventionally, prostanoids derived from COX-1 activity are associated with homeostatic cellular processes while COX-2 activity is more linked to pathologic prostanoid synthesis under inflammatory conditions. 3 COX-2 expression, however, is also necessary for homeostatic renal and cardiovascular function. 3 COX-2 synthesizes prostaglandin E2 and prostaglandin F2α in ureteral cells. Its increased expression results in nociception and smooth-muscle hypercontractility via paracrine signaling through neuronal and smooth muscle cells (SMCs), respectively. Nonsteroidal anti-inflammatory drugs (NSAIDs) can directly inhibit COX activity and thus reduce prostanoid synthesis, but clinical findings conversely show that COX-2–specific NSAIDs (coxibs) do not lower pain analog scores or increase stone passage rates when administered for obstructive urolithiasis. 4,5 The adverse cardiovascular and renal effects associated with administering NSAIDs during renal insufficiency also raise concerns about their chronic use as safe analgesics for UTO.
Despite the overwhelming biomechanical descriptions of pain generation during UTO, there are currently no pharmacologic therapies based on this process. Our previous work has consistently focused on deciphering the signaling pathways that regulate COX-2 expression during UTO. We have described previously calcium (Ca)2+- and protein kinase C (PKCζ)-dependent COX-2 expression in a physiologically relevant primary human urothelial cell (HUC) stretch model. 6 The present investigation uses a mouse UTO model and an in vitro urothelial cell stretch model to provide evidence that phosphatidylinositol 3-kinase (PI3K) mediates stretch-induced COX-2 expression in urothelial cells during UTO. PI3K is a mediator of several antioxidant responses, extracellular matrix remodeling, and cell proliferation processes activated by cell stretch. 7 –9
The following study provides preliminary evidence that inhibiting upstream mediators of stretch-induced COX-2 expression can provide pharmacologic targets of drug action for targeted therapeutics for symptomatic UTO.
Materials and Methods
Animals
Adult male CD-1 mice (12–14 weeks old, Charles River, Wilmington, MA) were housed under pathogen-free conditions in the University of Wisconsin-Madison School of Medicine and Public Health vivarium. Mice were housed in Innovive sterile ventilated microisolator cages (up to four per cage) under controlled ambient temperature (18°–26°C, 30%–70% humidity), and a 12-hour light/12-hour dark cycle. All mice were provided a standard rodent chow diet and sterile water ad libitum.
Technique for mouse unilateral ureteral obstruction
All procedures were conducted under approval and guidance of the Institutional Animal Care and Use Committee. Adult male CD-1 mice were anesthetized with 3% isoflurane and prepared for aseptic surgery. We identified the right ureter via midline abdominal incision and mobilized the ureter from surrounding fascia. The ureter was then obstructed via double-ligation at the ureterovesical junction with 6-0 nylon suture as described previously. 10 The unligated contralateral ureter was liberated through comparable methods and used as an internal sham operation.
A visual assessment of acute hydroureter after ligation, and hydronephrosis after treatment, were used as markers of a successful obstruction. We assessed and quantified the topologic changes to obstructed and contralateral ureters by recording the average longitudinal (12–6 o'clock) and the latitudinal (9–3 o'clock) radial measurements of cross-sectioned ureters. We observed a large increase in lumen area and an elongation of ureteral cells after 4 hours of obstruction compared with unobstructed contralateral tissues (Figs. 1A, 1B). Lumen diameter in mouse ureter obstructed for 4 hours increased an average of ∼3-fold (n=3, average longitude increase=22–60-microns, average latitude increase=12–50-microns). Contralateral ureters also displayed an average increase (∼2.5-fold) in lumen area (n=3, average longitude=15–35 microns, average latitude=7–20 microns) after 4 hours of obstruction. These observations were expected because of the shunting of urine flow to the contralateral kidney during unilateral ureteral obstruction (UUO).

Stretch-induced cyclooxygenase (COX)-2 expression in a unilateral uteteral obstruction (UUO) mouse model.
After ligation, the surgical incision was stitched and the mice were administered buprenorphine (0.1 mg/kg intraperitoneal) for postoperative analgesia, and treated with either vehicle or LY294002 for 1-hour before UUO. Mice were reanesthetized after treatment (time of obstruction), and the obstructed and unligated contralateral ureters were harvested above the ligation. Ureter samples were fixed with 10% formalin. Mice were sacrificed with 100% CO2 as approved by the American Association for the Assessment of Laboratory Animal Care.
Histology
After mouse ureters (obstructed and contralateral) were harvested, fixed specimens were routinely processed stepwise through graded ethanol (70%, 95%, 100% v/v), cleared in xylene, and embedded longitudinally in paraffin. Segments were serially cross-sectioned (5-micron), deparaffinized in xylene, and rehydrated through stepwise decreasing ethanol gradients (100%, 95%, 70% v/v). Samples were then subjected to hematoxylin and eosin (H&E), or Wright-Giemsa histology staining protocols. H&E staining was used to identify cellular nuclei (purple) and cytosol (pink). Wright-Giemsa staining highlights: Nuclei (purple), cytosol (various blues), collagen (pale pink), erythrocytes (bright pink), mast cells (bright purple), polymorphonuclear neutrophils (red granules, pink cytosol), eosinophils (red granules, blue cytosol), basophils (dark red granules, blue cytosol), monocytes (pale blue cytosol), lymphocytes (red granules, sky blue cytosol), and platelets (violet granules).
Immunohistochemistry (IHC)
Obstructed and contralateral mouse ureters were harvested, fixed with 10% formalin, and routinely processed stepwise through graded ethanol (70%, 95%, 100% v/v), cleared in xylene, and embedded longitudinally in paraffin. Segments were serially cross-sectioned (5-micron), deparaffinized in xylene, and rehydrated through stepwise decreasing ethanol gradients (100%, 95%, 70% v/v). Antigen retrieval (20 psi, 125°C, 2 min; 30 min 25°C) was performed in 10 mM sodium citrate buffer (pH 6.0) or with Biocare Decloaker® for anti-CD45 treatments (Biocare Medical, Concord, CA), and endogenous peroxidase activity was inactivated with 3% H2O2. Samples were washed with TBS, treated with blocking reagent (Background Sniper® Biocare Medical, Concord, CA) for 15 minutes, and incubated with either; antismooth muscle actin (BDbiosciences, San Jose, CA), antipan-cytokeratin (BDbiosciences, San Jose, CA), anti-COX-2 (Cell Signaling, Danvers, MA), or anti-leukocyte common antigen (LCA) (BDbiosciences, San Jose, CA) antibodies as described by the manufacturer. Samples were washed and incubated with antirabbit or antimouse secondary horseradish peroxidase (HRP)-conjugated antibody for 30 minutes, washed again, then incubated with 3,3′diaminobenzidine chromogen and rinsed with ddH20. Samples were visualized with a Nikon Eclipse 600® microscope, and the appearance of a brown precipitate indicated positive antibody staining. Images were recorded via Olympus Micro® software.
Protein isolation from ureteral segments
Harvested mouse ureteral segments were mechanically homogenized with a 1.5 motorized Pellet Pestle® (Kimble-Chase, Vineland, NJ) in homogenization buffer (100 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.8, 125 μL/ureter) for 30 minutes at 4°C. The resulting homogenate was centrifuged at 10,000×g for 15 minutes, and the protein-containing supernatant was collected. Protein concentration was determined via bicinchoninic acid (BCA) colorimetric assay (Thermo Scientific, Rockford, IL) for further analysis.
Western blotting
Cell lysates or tissue homogenates were subject to BCA assay for protein quantification, and resolved via sodium dodecyl sulfate polyacrylamide gel electrophoresis (4%–20%, 15 μg/well). Proteins were transferred to nitrocellulose blotting membranes (Biorad Laboratories, Hercules, CA) and incubated overnight in blocking solution (1% milk, 1% bovine serum albumin, 0.1% v/v phosphate-buffered saline-Tween20 (PBS-Tween). Blots were incubated with specific antibodies overnight, washed three times with PBS-Tween (0.1% v/v), and incubated with HRP-conjugated secondary antibodies for 1 hour. The resulting samples were washed twice with PBS-Tween, rinsed with PBS, and developed via chemiluminescence with SuperSignal West-Femto® (Thermo Scientific, Rockford, IL). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) protein expression was used as a loading control for all Western blots. The resulting blots were converted into digital images via HP Scanjet 4370® and quantified by densitometry with Adobe Photoshop CS.® GAPDH monoclonal antibody was purchased from Abcam (Cambridge, MA), and COX-2 monoclonal antibody was purchased from Cayman Chemical (Ann Arbor, MI). Antibodies for P110α (#4254), P110β (#3011), PDK-1 (#3062), pT410-PKCζ (#9378), PKCζ (#9372) were purchased from Cell Signaling Technology (Danvers, MA), and antibodies for P110γ (sc-166365) and P110δ (sc-55589) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Primary HUC culture
All procedures were conducted under approval and guidance of the Institutional Review Board. Normal human ureteral segments were collected from healthy patients undergoing nephrectomy for kidney donation and transported to our laboratory in Ham F-12 media. Normal primary HUCs were cultured (5% CO2, 37°C) from ureter specimen as optimized by our collaborators.
11
Briefly, ureters were opened to expose the urothelium, and the urothelial layer was manually removed and minced. The minced tissue was transferred to collagen I-coated sterile tissue culture plates and cultured for 72-hours in Ham F-12 (Sigma-Aldrich, St. Louis, MO) supplemented with 5% (v/v) fetal bovine serum (FBS, Sigma), 5 μg/mL apo-transferrin (Sigma), 2.7 mg/mL glucose (Sigma), 0.1 mM nonessential amino acids (Sigma), 100 U/mL penicillin, 100 μg/ml streptomycin (Sigma), and 2 mM
Primary HUC stretch
After being cultured from normal human ureter, HUCs were passaged four times and cultured on collagen I-coated Bioflex® stretch plates (FlexCell Int. Corp, Hillsborough, NC). HUCs on stretch plates were cultured to >90% confluency and cyclically stretched via the FX-3000T Flexercell apparatus. Cyclic stretch was performed with 5% to 20% displacement at 12-cycles/min as optimized previously for in vitro stretch-induced COX-2 expression in HUCs. 6 After stretch, cells were lysed in “lysis buffer” (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 10 μg/mL each of leupeptin, aprotinin, antipain, and 1% TritonX-100), and the protein-containing supernatant was collected for further analysis.
Pharmacologic inhibition of PI3K in HUCs
The vehicle for LY294002 was dimethyl sulfoxide (DMSO), where HUC treatments contained a final concentration of 0.01% DMSO v/v in supplemented Hams F-12 media. LY294002 doses were not cytotoxic to epithelial cell cultures as previously reported. 12,13 HUCs were also assessed via gross morphology and did not display any apparent signs of cytotoxicity.
RNA-interference
HUCs were cultured to 50% confluency on collagen I-coated Bioflex stretch plates as described above. 6 Oligofectamine™-siRNA complexes (Invitrogen, Carlsbad, CA) were generated in Ham F-12 media according to manufacturer recommendations using siRNA oligonucleotides targeted against specific mRNA sequences (PI3Kα=GUGGUAAAGUUCCCAGAUA, PI3Kβ=GAUUAUGUGUUGCAAGUCA, PDK-1=GAAGCAGGCUGGCGGAAACUU; Dharmacon, Chicago, IL), or nontargeting mRNA sequences (ONTargetplus #1-D-001810-01-05; Dharmacon, Chicago, IL). Oligofectamine™-siRNA complexes were added to HUC cultures and incubated for 4 hours at 37°C (5% CO2, 95% air). The final concentration of siRNA oligonucleotide in each well was 50 nM. After the initial 4-hour incubation period, the medium was replaced with supplemented (5% FBS) Ham F-12 medium every 48 hours. After an optimal 72-hour mRNA knockdown period was concluded, HUCs were cyclically stretched for 6 hours as described above, and protein extracts were analyzed by Western blotting.
Statistical analyses
Statistical significance was qualified by unpaired Student t test. Statistical significance was defined as P value≤0.05 unless otherwise stated. All histograms are expressed as mean±standard error of the mean.
Results
A histologic analysis of cell stretch during UUO
UUO stretched urothelial cells and caused an average ∼5-fold decrease (n=3, average longitude urothelium decrease=116–22 microns, average latitude urothelium decrease=126–25 microns) in urothelium thickness. SMC layer thickness also decreased ∼2.5-fold (n=3, average longitude=39–20 microns, average latitude=53–18 microns) because of cell stretch during UUO. Contralateral urothelial cells stretched and reduced urothelial cell layer thickness (1.5-fold, average longitude=10–6 microns, average latitude=14–6 microns), and SMC layer thickness (1.5-fold, average longitude=7–4 microns, average latitude=8–5 microns). These results indicate that cell stretch occurs in urothelial cells during UUO.
Evidence for stretch-induced COX-2 expression during UUO
After confirming cell stretch during UUO, we used IHC to visualize cell types associated with COX-2 expression (Fig. 1C). Unobstructed (0-hour) ureters displayed COX-2 expression along the apical surface of urothelial cells with COX-2 expression also present in SMCs. Ureters obstructed for 2 hours showed higher COX-2 expression in urothelial cells compared with SMCs. Mouse ureters obstructed for 4 hours had strong chromogen staining in urothelial cells. SMCs in ureters obstructed for 4 hours also displayed COX-2 expression but were not comparable to urothelial cell COX-2 expression. Also, we observed an increase in urothelial cell COX-2 expression in contralateral ureters within 2 hours of obstruction; however, we did not observe further increases in COX-2 expression at 4-hour time points (Fig. 1D).
The classic inflammatory cascades that induce COX-2 expression are cytokine-mediated responses to infiltrating leukocytes that migrate into damaged renal tissues between 4 and 12 hours after obstruction. 14 We used both Wright-Giemsa staining and specific antibodies for LCA/CD45, an antigen present on all leukocyte cell types, to determine whether leukocyte migration into obstructed ureteral tissues occurs in concert with maximal COX-2 expression at 4 hours of UUO. Wright-Giemsa staining shows that neutrophils, eosinophils, basophils, and lymphocytes are not present within 4 hours of obstruction (Fig. 1E).
We also did not observe a significant amount of mast cells present in mouse ureters at any time point. LCA/CD45 staining confirmed the results from Wright-Giesma stains. Mouse ureters obstructed for 0, 2, or 4 hours had no visible staining for LCA/CD45 positive cell populations under our conditions (Fig. 1F).
Therefore, the collective results support stretch-induced COX-2 expression as the primary mechanism of increased COX-2 expression in urothelial cells during UUO. This mechanism is shared with human obstructive uropathy. 6
PI3K mediates in vivo stretch-induced COX-2 expression
We identified PI3K-dependent stretch-induced COX-2 expression in urothelial cells. Mice were dosed via intraperitoneal injections of 25% DMSO/PBS (vehicle), PI3K inhibitor LY294002 (3, 30 mg/kg) (IC50=1.4 μM, Calbiochem, Gibbstown, NJ) to evaluate the effects of pharmacologic PI3K inhibition on in vivo stretch-induced COX-2 expression.
Vehicle-treated mice displayed intense COX-2 staining in the urothelial cells and SMCs of obstructed ureters (Fig. 2A). Vehicle-treated contralateral ureters expectedly did not display significant COX-2 expression as assessed by IHC. LY294002 (3mg/kg)-treated samples displayed intense urothelial cell COX-2 staining that was comparable to vehicle-treated obstructed ureters but had less intense COX-2 staining in their SMCs (Fig. 2B). The contralateral ureters of mice treated with 3 mg/kg LY294002 also had more intense COX-2 staining in urothelial cells and SMCs compared with vehicle-treated contralateral ureters (Fig. 2B). Notably, no significant urothelial cell COX-2 expression was observed in obstructed or contralateral ureters in mice administered 30mg/kg LY294002 (Fig. 2C). SMCs in obstructed ureters, however, were positive for COX-2 expression.
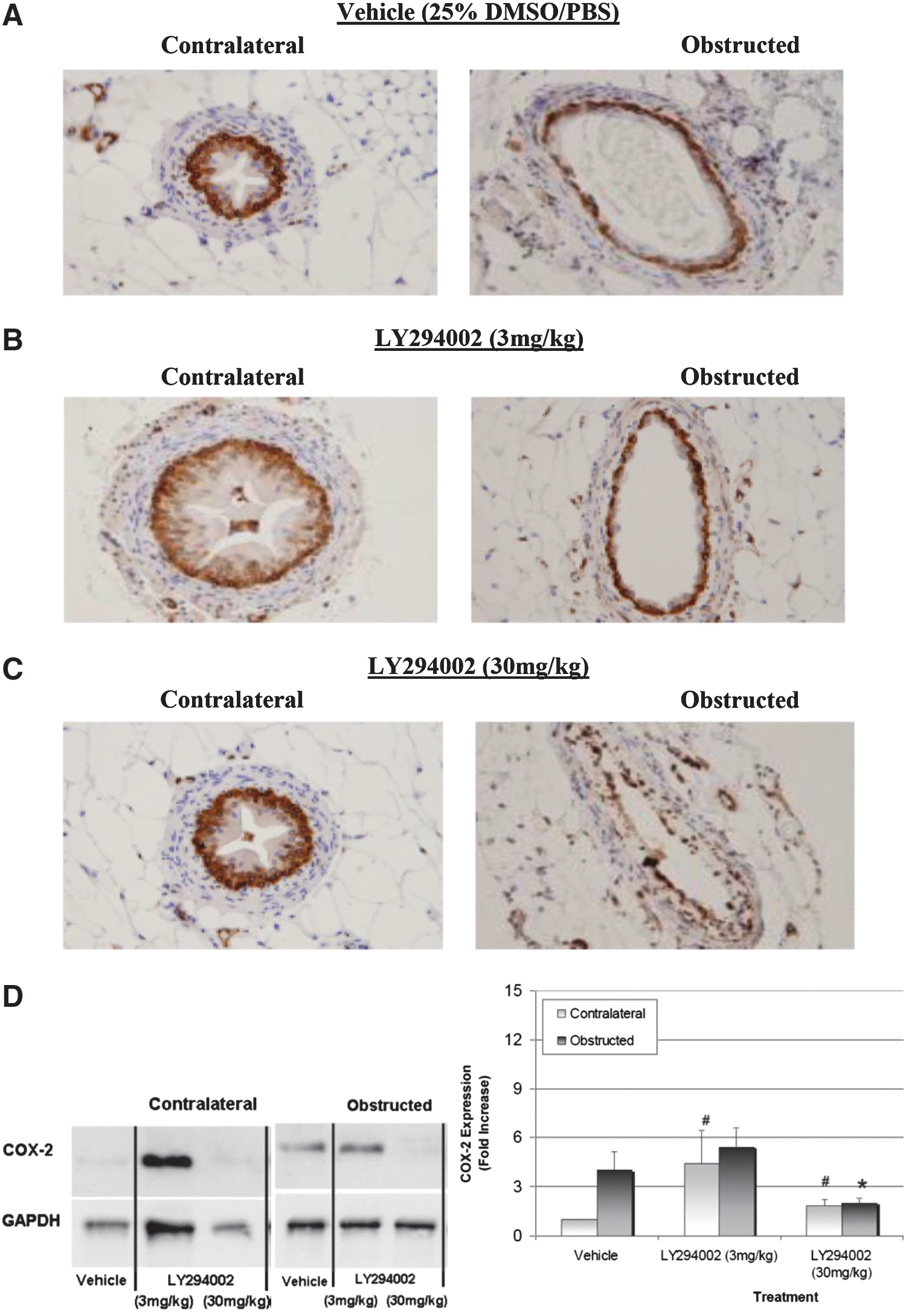
Phosphatidylinositol 3-kinase mediates stretch-induced cyclooxygenase (COX)-2 expression during unilateral ureteral obstruction (UUO). Mice were administered either dimethyl sulfoxide/phosphate buffered saline (DMSO/PBS) (25% v/v) or LY294002 (3 mg/kg, 30 mg/kg) 1 hour before UUO. Mouse ureters were obstructed for 4 hours, fixed, and probed with specific antibodies for COX-2 to visualize with chromogen (brown) staining (n=3).
After IHC showed that administering LY294002 can attenuate in vivo stretch-induced COX-2 expression, Western blotting was performed to evaluate quantitatively the effects of LY294002 administration on stretch-induced COX-2 expression (Fig. 2D). UUO induced a 4-fold increase in COX-2 expression in vehicle-treated obstructed mouse ureters (Fig. 2D). LY294002 (3 mg/kg) caused a 5-fold (P=0.09) increase in COX-2 expression in contralateral ureters, while only a 2-fold (P=0.06) increase was observed at the higher dose of 30 mg/kg. LY294002 (3 mg/kg) did not attenuate stretch-induced COX-2 expression, but administering 30 mg/kg significantly reduced stretch-induced COX-2 expression in obstructed mouse ureters by 50% (P=0.09) compared with vehicle-treated mice.
PI3K mediates in vitro stretch-induced COX-2 expression
HUCs were cultured on six-well stretch plates and preincubated with 0, 30, and 100 μM doses of LY294002 for 1 hour before cyclic stretch. A representative Western blot shows cyclic stretch caused an average 15-fold increase in COX-2 expression and was reduced by LY294002 (70%, 30 μM, P=0.019) compared with COX-2 expression in stretched untreated cells (Fig. 3A).
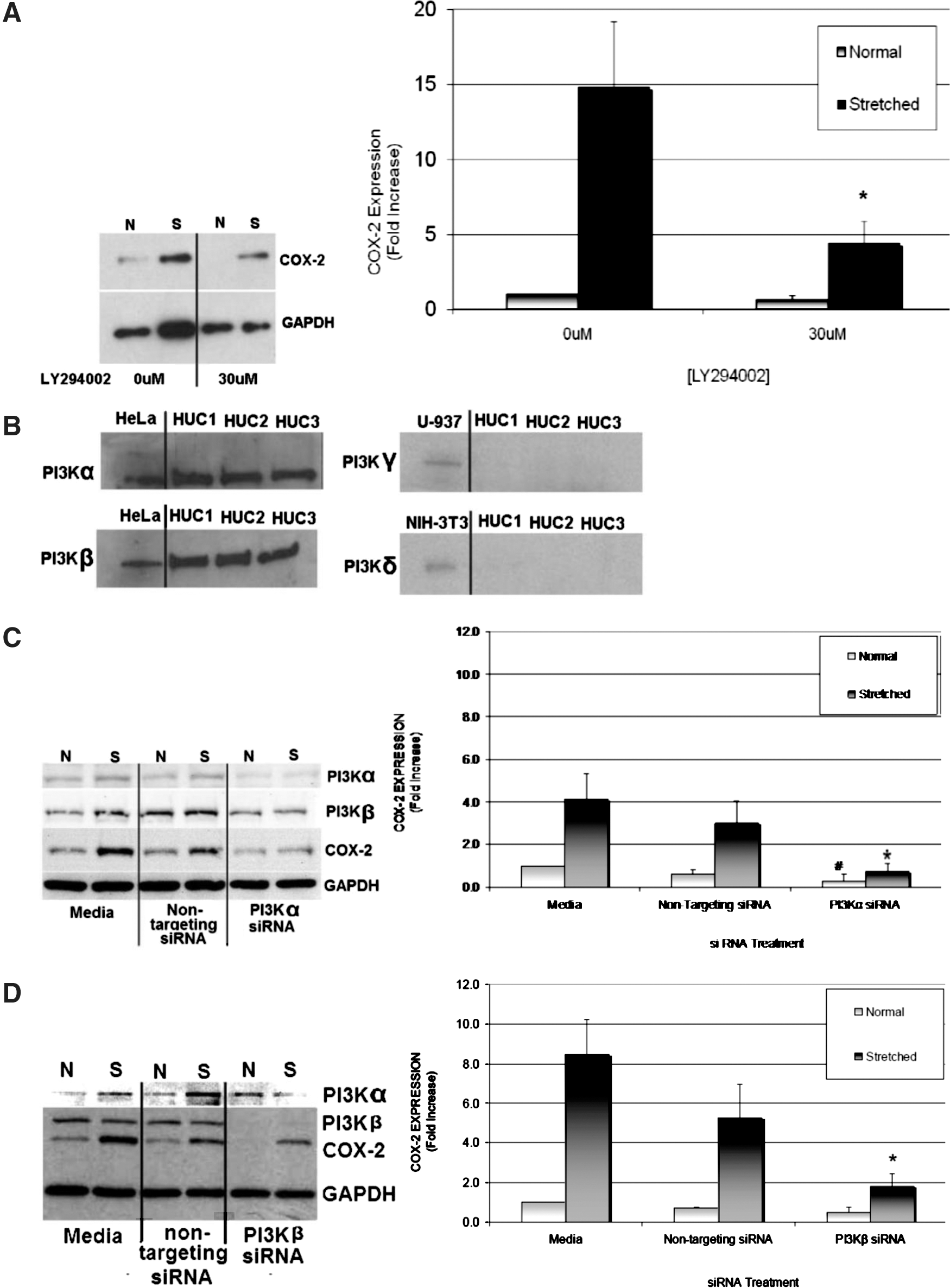
Phosphatidylinositol 3-kinase (PI3K) mediates stretch-induced cyclooxygenase (COX)-2 expression in human urothelial cells (HUCs). HUCs were preincubated with LY294002 (30 μM) for 1 hour before cyclic stretch for 6 hours. Cell lysates were resolved via Western blotting, and specific antibodies were used to assess COX-2 expression in normal (N) and stretched (S) HUCs.
Given that LY294002 showed efficacy for attenuating stretch-induced COX-2 expression, we corroborated the pharmacologic results with RNA-interference. First, we determined which PI3K isoforms were expressed in HUCs in our preparations. HeLa (human epithelial), U-937 (human monocyte), and NIH-3T3 (mouse fibroblast) were used as control cell types for PI3Kα/β, PI3Kγ, or PI3Kδ expression, respectively. Western blotting with specific antibodies for PI3K isoforms identified PI3Kα and PI3Kβ as the predominant PI3K isoforms expressed in HUCs (Fig. 3B), while PI3Kγ and PI3Kδ protein expression was not detected in our HUC preparations.
PI3Kα- or PI3Kβ-targeting siRNA were used to specifically attenuate PI3Kα and PI3Kβ expression, respectively. RNA-interference via siRNA reduced PI3Kα expression in HUCs by ≥88% and PI3Kβ expression by ≥99% compared with their normal protein expression levels. HUC treatments with specific siRNA directed against PI3Kα showed a significant reduction in PI3Kα and COX-2 expression without altering PI3Kβ expression and vice versa (Fig. 3C).
PI3Kα-siRNA–treated cells displayed an average 82% (P=0.02) decrease in stretch-induced COX-2 expression and an average 70% (P=0.02) decrease in basal COX-2 expression (Fig. 3C). HUCs treated with PI3Kβ-siRNA before cyclic stretch displayed an average 80% (P=0.01) reduction in stretch-induced COX-2 expression, but basal COX-2 expression was not altered (Fig. 3D).
Stretch-induced PKCζ activation is PI3K-dependent
Activating the ζ isoform of PKC depends on phosphoinositide 3,4,5-triphosphate (PIP3) synthesized by PI3K at cellular membranes. 15 We reported previously that stretch activates PKCζ upstream of stretch-induced COX-2 expression. 6 In the present study, we sought to identify a role for PI3K in stretch-induced PKCζ activation by Western blotting for the presence of pT410-PKCζ, a phosphorylation site necessary for PKCζ activation. 15 A 5-fold increase (P=0.01) in PKCζ activation occurs within 2 hours of HUC stretch, and PKCζ expression also appeared to increase 1.5-fold (P=0.03) within 1 hour of HUC stretch (Fig. 4A).
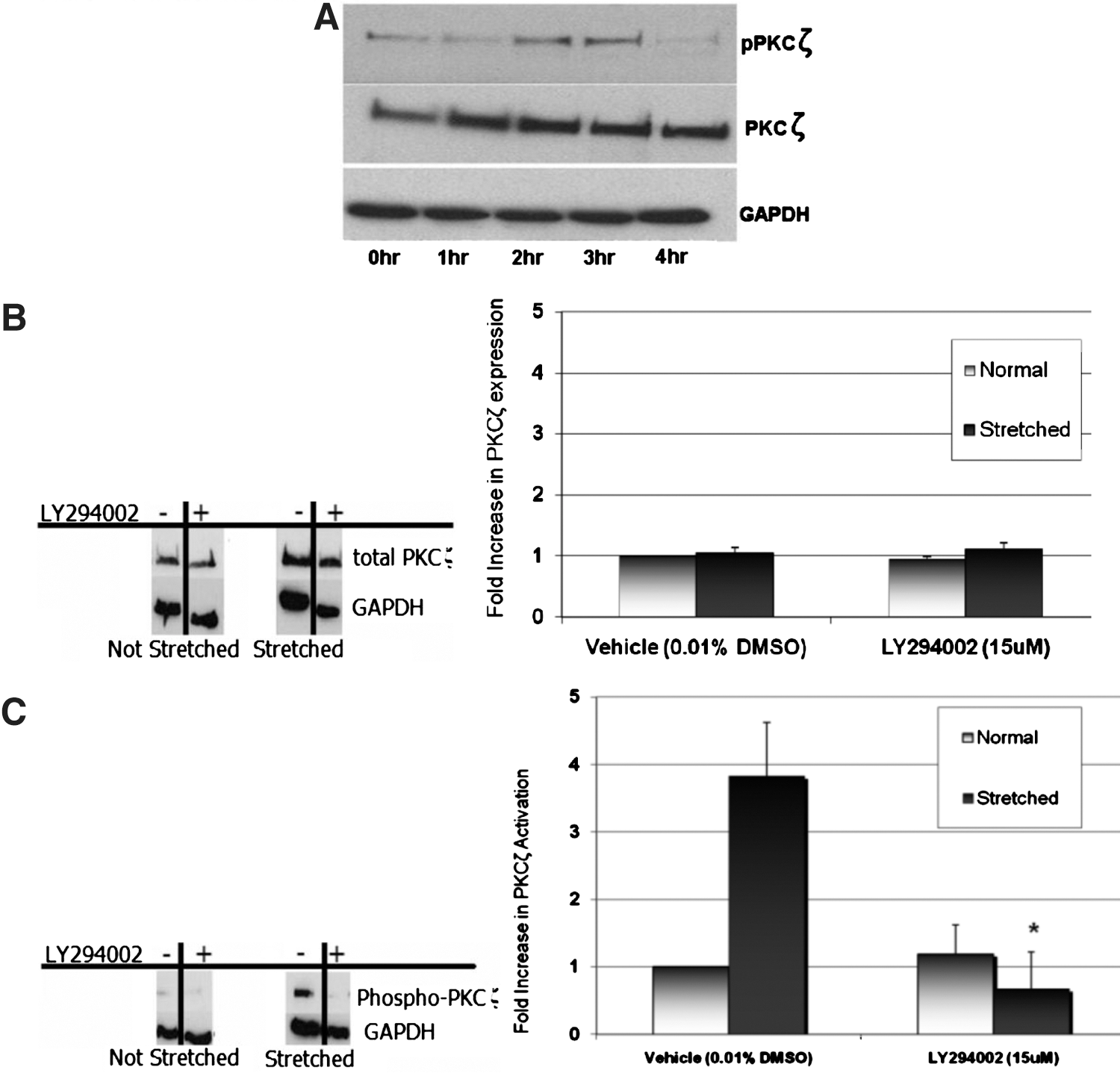
Phosphatidylinositol 3-kinase (PI3K) mediates stretch-induced protein kinase C (PKCζ) activation in human urothelial cells (HUCs).
PI3K inhibitor LY294002 was used to identify PI3K-dependent stretch-induced PKCζ activation. Figure 4B shows a representative Western blot of the effects of pharmacologic PI3K inhibition on the presence of total PKCζ. The results show that administering LY294002 during HUC stretch does not alter PKCζ expression but does cause an 82% decrease (P=0.007) in stretch-induced PKCζ activation (Fig. 4C). These results indicate that both stretch-induced PKCζ activation and COX-2 expression are PI3K-dependent.
Discussion
The results of this study indicate that PI3K-dependent stretch-induced COX-2 expression occurs during UTO. Our in vivo findings are therefore physiologically comparable to leukocyte-independent mechanisms of COX-2 expression observed during human UTO. 16 We further showed that PI3K signaling is upstream of both stretch-induced PKCζ activation and stretch-induced COX-2 expression during urothelial cell stretch.
Stretch-induced COX-2 expression is important for several homeostatic functions including bone density maintenance, bladder filling and voiding, pregnancy and labor, and vascular endothelial cell function. Unfortunately, stretch-induced COX-2 expression is also central to preterm births from multiple pregnancies, ventilator-induced lung injury, bone density loss in microgravity, and obstructive uropathy.
UUO mouse models have been used routinely to investigate renal pathology during hydronephrosis. Consistent with the findings from other various species, we reported previously that UTO in an acute (<4 hours) UUO mouse model displayed increased COX-2 expression. 10,16 Thus, a UUO mouse model provides a readily available in vivo method to histologically characterize stretch-induced COX-2 expression in different cell types during UUO.
We identified COX-2 expression before a typical time course of leukocyte-mediated COX-2 expression (12 hours) and provided evidence for in vivo stretch-induced COX-2 expression during acute UTO. 14 Although we previously showed maximal COX-2 expression within 4 hours of UTO, whether pathologic COX-2 expression in chronically obstructed ureters is maintained by infiltrating leukocytes in a mouse model was not addressed by this study. Our previous analyses of chronically obstructed human ureter, however, did not present any evidence of immune cell infiltration and therefore does not predict an alternative process driven by leukocytes.
PI3K inhibitor LY294002 has been used previously to inhibit in vivo PI3K-dependent functions such as ovarian cancer cell growth at dosages up to 100 mg/kg/d. 17 We demonstrate LY294002 efficacy for reducing in vivo stretch-induced COX-2 expression at 30 mg/kg in a UUO mouse model. The 3 mg/kg dose of LY294002, however, unexpectedly caused a 5-fold increase in both obstructed and contralateral COX-2 expression during UTO. Because we did not observe a comparable induction of COX-2 expression in the in vitro HUC stretch model, determining the effects of paracrine signaling from stretched SMCs or resident fibroblasts may provide the information necessary to resolve the inconsistency.
Whether COX-2 expression is stimulated similarly in SMCs and urothelial cells also remains an interesting question. LY294002 showed little efficacy for attenuating increased COX-2 expression in SMCs compared with urothelial cells in the UUO mouse model. This may be because of innate variables in the model such as LY294002 tissue distribution in the ureter, and the presence of COX-2 expression pathways that are not dependent on PI3K. We suggest that the most plausible explanation for this observation is that LY294002 has less efficacy SMCs compared with urothelial cells because SMCs stretch less than urothelial cells under our experimental conditions. Hence, the increase in COX-2 expression observed in SMCs during UUO could either be the direct result of SMC stretch or the indirect result of paracrine signaling from stretched neighboring cell types (ie, fibroblast, urothelial cells). Determining the mechanisms of increased COX-2 expression in SMCs, however, was outside the scope of this study, which focused on the primary site of COX-2 expression—urothelial cells.
Notably, PI3K inhibition did not reduce COX-2 expression below homeostatic levels. In terms of therapeutic application, this attribute may circumvent NSAID toxicity associated with inhibiting homeostatic COX-2 activity in the urinary tract during renal insufficiency. Chuang and colleagues 18 reported that selective COX-2 inhibitor celecoxib can attenuate COX-2 expression and thus reduce tissue damage during chronic UTO (>12 days). However, the human clinical studies performed by Phillips and associates 4 using celecoxib showed no analgesic properties for selective COX-2 inhibition during obstructive stone disease. Our results confirm that PI3K is involved in the initiation of stretch-induced COX-2 expression, but whether PI3K inhibitors can reduce stretch-induced COX-2 expression after it has occurred was not addressed by the current experimental design. Therefore, conclusions about the therapeutic efficacy of LY294002 for obstructive uropathy remain limited.
There is a growing interest in isoform-specific PI3K inhibitors for anti-inflammatory use. Our results reveal a possible distinction between the roles of PI3Kα and PI3Kβ in stretch-induced COX-2 expression as assessed by RNA-interference. Attenuating PI3Kα expression inhibited both basal and stretch-induced COX-2 expression in HUCs, but modulating PI3Kβ expression specifically reduced stretch-induced COX-2 expression. Nonredundant roles for PI3Kα and PI3Kβ have been described previously in carcinogenesis. 19,20 Our current study suggests that PI3Kα and PI3Kβ may also have distinct roles in stretch-induced COX-2 expression.
PI3K is upstream of PKCζ activation in response to several cytokines and growth factors, and regulates COX-2 expression through its post-transcriptional control of COX-2 mRNA stability. 15,21 –23 Our study demonstrates that PI3K is also upstream of PKCζ activation in response to urothelial cell stretch, as stretch-induced PKCζ activation is sensitive to PI3K inhibition under our conditions. Because PKCζ activation routinely depends on PI3K, our observations of PI3K-dependent PKCζ activation during urothelial cell stretch were plausible. Nonetheless, to our knowledge, this is the first investigation to identify PI3K-dependent PKCζ activation in response to a mechanical stimulus. PKCζ is upstream of NF-κB and mitogen activated protein kinase signaling cascades, both of which have demonstrated the ability to regulate COX-2 expression and stimulate proinflammatory gene expression. 15 More importantly, PKCζ regulates hnRNPA1, which can bind and stabilize AU-rich proinflammatory mRNA transcripts such as COX-2. 24
We previously reported that COX-2 expression in HUCs is primarily regulated via post-transcriptional mechanisms involving increased COX-2 mRNA stability. 16 Therefore, we further hypothesize that PI3K-dependent PKCζ activation may be involved in stabilizing COX-2 mRNA during cell stretch and increasing its protein expression.
Conclusions
These are the first descriptions of PI3K-dependent COX-2 expression in response to urothelial cell stretch. Despite the limited use of NSAIDs for obstructive uropathy, suppressing pathologic COX-2 activity remains a laudable end point for the effective medical management of UTO. There are currently no targeted therapies for obstructive uropathy. We propose that therapeutics designed to reduce COX-2 activity during UTO can be developed based on urothelial cell responses to stretch.
Footnotes
Acknowledgments
We wish to thank Dr. Walter Hopkins for surgical and laboratory equipment, Dr. Arjang Djamali and Drew Roenneburg for histology resources, Kim Mauer for surgical procedure support, and Dr. Travis Jerde for his expertise.
The David T. Uehling Professorship at the University of Wisconsin-Madison Department of Urology is supported by a grant.
Disclosure Statement
No competing financial interests exist.