
Abstract
Background and Purpose:
Magnesium (Mg2+) has been shown to be a kidney stone inhibitor; however, the exact mechanism of its effect is unknown. Using theoretical models, the interactions of calcium and oxalate were examined in the presence of Mg2+.
Methods:
Molecular dynamics simulations were performed with NAMD and CHARMM27 force field. The interaction between calcium (Ca2+) and oxalate (Ox2−) ions was examined with and without magnesium. Concentrations of calcium and oxalate were 0.1 M and 0.03 M, respectively, and placed in a cubic box of length ∼115 Angstrom. Na+ and Cl− ions were inserted to meet system electroneutrality. Mg2+ was then placed into the box at physiologic concentrations and the interaction between calcium and oxalate was observed. In addition, the effect of citrate and pH were examined in regard to the effect of Mg2+ inhibition. Each system was allowed to run until a stable crystalline structure was formed.
Results:
The presence of Mg2+ reduces the average size of the calcium oxalate and calcium phosphate aggregates. This effect is found to be Mg2+ concentration-dependent. It is also found that Mg2+ inhibition is synergistic with citrate and continues to be effective at acidic pH levels.
Conclusion:
The presence of magnesium ions tends to destabilize calcium oxalate ion pairs and reduce the size of their aggregates. Mg2+ inhibitory effect is synergistic with citrate and remains effective in acidic environments. Further studies are needed to see if this can be applied to in vivo models as well as extending this to other stone inhibitors and promoters.
Introduction
T
Methods
Using the NAMD program, molecular dynamics (MD) simulations were performed to evaluate the role of Mg2+ in calcium and oxalate binding. 5 NAMD is a molecular dynamics code used in the examination of large systems, including biologic systems. Using CHARMM potential functions, NAMD describes individual particles at a molecular level with force field specifications. 5 All ions studied used the CHARMM27 force field. 6,7 Force field parameters determined by Yesselman and associates 8 via MATCH were used for Ox2− and citrate (Cit3−), while those for phosphate (PO4 3−) and dihydrogen phosphate (H2PO4 −) were determined using CHARMM general force field (CGenFF) 9 in the CHARMM program. The standard TIP3P potential model was used for water. 10 The short-range repulsion and dispersion (Lennard-Jones interactions) and long-range Coulombic forces were evaluated and the Newton equation of motion was integrated to propagate the dynamics of the system.
Table 1 outlines the components of each system design. To avoid any confusion, we mention that size of systems 1 and 2 is small; specifically, they are 15 times smaller than systems 4 to 10. We used systems 1 and 2 to simply evaluate the interaction of Ca2+ and Ox2− in the absence and presence of Mg2+ via the free energy calculations. System 1 consists of one pair of Ca2+ and Ox2− and 3330 water molecules. In addition, 10 pairs (∼0.16 M) of Na+ and Cl− ions were added, so that its overall salt concentration is close to the physiologic condition. Compared with system 1, one Mg2+ and two additional Cl− ions were added to system 2. The latter is to meet the system electroneutrality. The levels of the ions were supraphysiologic to observe any effect within the constraints of the computing power. After equilibration, we computed free energies of systems 1 and 2, using the metadynamics algorithm.
11
Two coordinates, ">
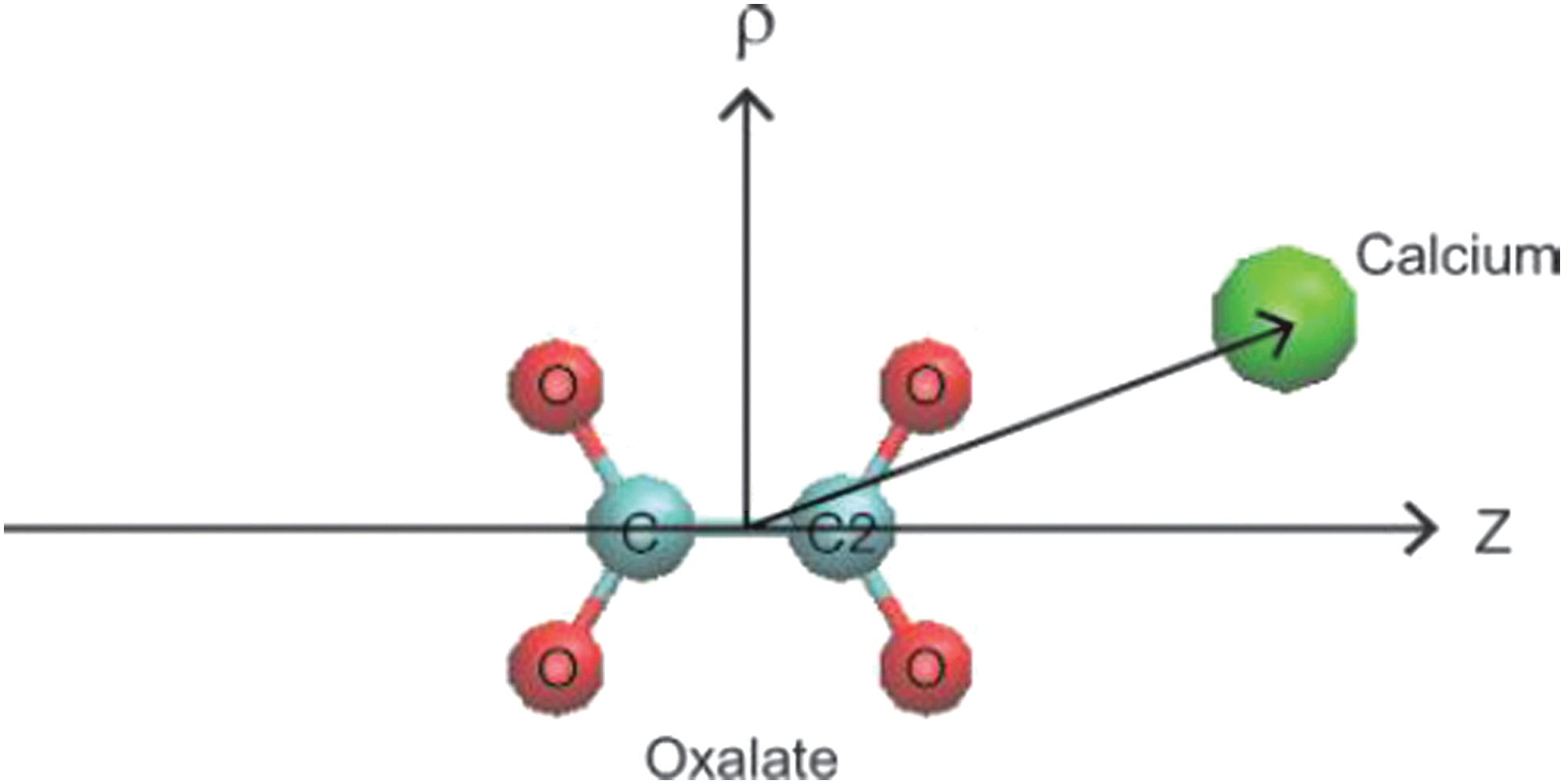
Coordinates of calcium and oxalate: ρ and z are defined as the displacement of Ca2+ from the Ox2− center-of-mass along the carbon–carbon bond in Ox2− and its perpendicular directions, respectively. Ca2+ ion is represented in the green sphere and O and C atoms of Ox2− are in red and light blue, respectively. Visual Molecular Ddynamics 18 is used for display.
The free energy results for Ca2+ and Ox2−, in particular, local minima, provide information on the Ca2+-Ox2− binding affinity and structures and their variations with the presence of other ions. Deeper energy minima indicate the formation of a more stable calcium-oxalate ion pair, thereby suggesting that calcium and oxalate ions are more likely to aggregate. Within our model, we are able to determine both the value and location of the free energy minima. We should note, however, that the effect of many urinary ionic species as well as that of heterogeneous biologic environments is ignored in the free energy calculations.
With systems 3 to 10, we investigated aggregation dynamics of CaOx in the absence and presence of various urine ions that are either known stone promoters or inhibitors of low weight. 12 –17 System 3 was composed of 22 pairs of Ca2+ and Ox2− ions, 30 pairs of Na+ and Cl− ions, and 10000 water molecules. For systems 4 to 10, 90 Ca2+ and 30 Ox2− were immersed in water consisting of 50,000 water molecules. The salt concentration was adjusted to satisfy the system electroneutrality. Systems 4, 5, and 6 that model a pH neutral/basic environment differ in their magnesium concentration; ie, 0, 83, and 150 mM, respectively (Table 1). For systems 7 and 8, citrate was inserted to evaluate its effect on aggregates. Systems 9 and 10 were generated from systems 4 and 6 by replacing their PO4 3− ions with H2PO4 − to model an acidic urine environment. This allows us to study how pH influences Ca2+-Ox2− aggregation. 100 ns, 80 ns and 40 ns MD simulations were performed for system 3, systems 4 to 8, and systems 9 and 10, respectively.
Results
In system 3, a large aggregate was formed around 30 ns. This structure continued to grow and become stabilized through association and dissociation of Ca2+ and Ox2− ions along its edges. After ∼50 ns, a stable structure was formed, and no significant dissociation or association along its edges was observed in the remainder of the simulation. The structure of the aggregate at 100 ns is shown in Figure 2. Its analysis reveals three important configurations for the binding pair. Specifically, Ca2+ ions bind to the O-C-O (b1) or the O-C-C-O (b2) pockets of the Ox2− anion. Oxygen atoms (b3) of oxalate can also function as a third cation binding site to form an ion pair with Ca2+. 8

Structure of the calcium-oxalate aggregate obtained from the 100 ns MD simulation of system 3. Ca2+ is represented in the green sphere and oxygen and carbon atoms in Ox2− are in red and blue spheres, respectively. Visual Molecular Dynamics was used to display the structure. 18
The free energy of a Ca2+ and Ox2− complex determined as a function of ρ and z shows that its global minimum is located at the b2 position (viz., ρ∼2.8Å and z=0 Å) of oxalate (Fig. 3A). This means that b2 is the strongest Ca2+ binding site of Ox2− when no promoter and/or inhibitor ions are present in the system. Local minima at ρ=0Å and z=∼±3 Å and at ρ∼3.2Å and z=∼±2 Å represent the second and third most stable binding sites, corresponding to b1 and b3, respectively.

Two-dimensional free energy map for the Ca2+-Ox2− complex in an aqueous solution of NaCl:
Compared with system 1, system 2 shows a slight decrease in the Ca2+ and Ox2− binding affinity because of the presence of Mg2+ (Fig. 3B). Mg2+ initially placed near the b2 site binds to the site during the equilibration (Fig. 3B). In the ensuing 400 ns simulation, it tends to move from one binding site to another and hinder the binding of Ca2+. Because Mg2+ is smaller, it can “fit” into the binding sites of Ox2− better than Ca2+. This can prevent the binding of Ca2+ and Ox2− at least at the Mg2+ occupied sites and, as a result, slow down the aggregation of Ca2+ and Ox2−. This paints the picture that Mg2+ and Ca2+ would compete directly for binding sites on Ox2−. The presence of shallow secondary free energy minima for Mg2+- Ox2− complexation, however, complicates this picture. As Mg2+ ions approach Ox2− from a distance, they can be trapped in these shallow wells. This could decelerate the binding of magnesium and considerably lower its ability to compete with Ca2+ to occupy the binding sites of Ox2−. To obtain further insight into this issue and the roles of Mg2+, we studied the formation of aggregates involving calcium explicitly via MD using systems 4 to 10 (Table 1).
Figure 4 shows the aggregate structures obtained for systems 4 to 6. We notice that Ca2+ form aggregates mainly with PO4 3− because of the strong Coulombic interaction of the latter. As a result, aggregates are primarily made up of Ca2+- PO4 3− (CaP) binding structure mixed with some CaOx structure (Fig. 4A). The most salient aspect of Figure 4 is that the size of CaOx/CaP aggregates generally decreases as the Mg2+ concentration is raised. As shown in Figure 4B–D, however, the difference in the aggregate size between the 0 mM (system 4) to 83 mM (system 5) cases is not significant, compared with the difference of systems 5 (83 mM) and 6 (150 mM). These results indicate that Mg2+ has an inhibitory effect on CaOx/CaP aggregation, but this effect is magnesium concentration-dependent.

Comparison of Ca2+ and Ox2− aggregate in relation to Mg2+concentration. In
Systems 7 and 8 evaluated the role of citrate in inhibition in the absence and presence of magnesium. Comparison of the results in Figure 5A and B suggests that citrate alone does not have a significant influence on the size of CaP aggregates. We speculate that citrate does not compete with PO4 3− because PO4 3− presents in higher concentration than citrate in the system, although they have the same valency. Because the aggregation is mainly effected via PO4 3− in the present case, citrate therefore tends to show little effect on CaP. The addition of citrate alone, however, can have an inhibitory effect on the formation of CaOx aggregates by directly competing with Ox2− for binding Ca2+, which is consistent with our previous free energy result. The role of citrate more clearly appears when both citrate and Mg2+ are present (Figs. 5C and D). Interestingly, when citrate was added with Mg2+, a synergistic effect was seen, as manifested by the small size of CaOx/CaP aggregates in system 8 (Fig. 5C).
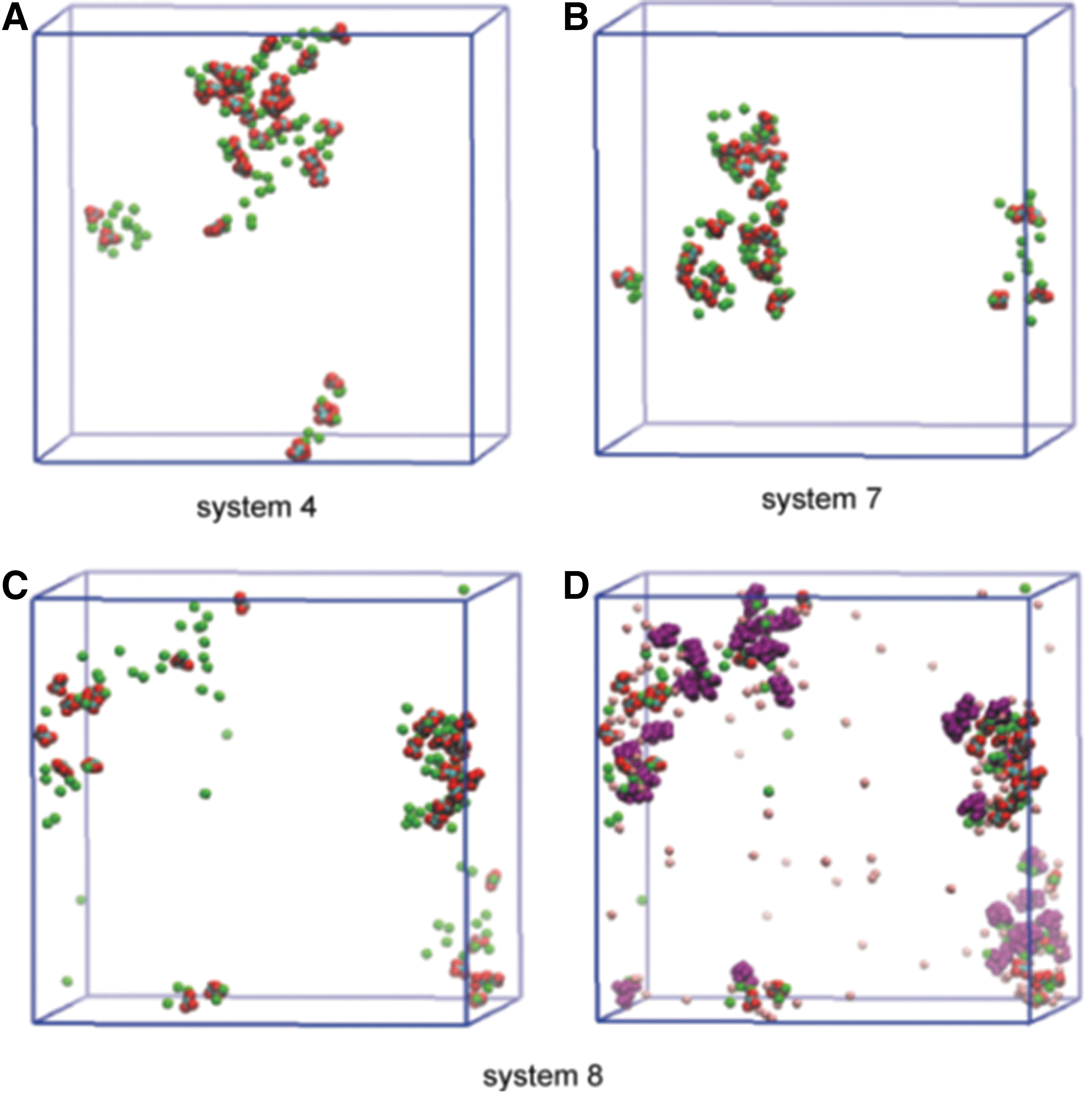
Effect of Mg2+ and Cit3−. In
Finally, the effect of urinary pH and Mg2+ was explored in systems 9 and 10 (Figs. 6B and C). H2PO4 − was used, which would be seen in a weakly acidic urine, as opposed to PO4 3−, which would be seen in the more pH neutral and/or basic environment. In this sense, the replacement of PO4 3− by H2PO4 − effectively represents lowering of pH. This results in the formation of mainly CaOx aggregates, because Ca2+ ions are no longer intercepted by trivalent phosphate and thus become more available to interact with Ox2− ions. This demonstrates the expected results of increase in CaOx aggregates in acidic urine. When Mg2+ was added to the system, the inhibitory effect as previously shown was maintained within this acidic milieu (Fig. 6C). We should note that Mg2+ ions on average do not actively participate in aggregation; ie, in most cases, they do not bind tightly to Ox2−. This seems to support our earlier view that Mg2+ screens the interactions of Ca2+ with Ox2− rather than directly competing with Ca2+ for the binding sites on Ox2−.

Effect of pH. All snapshots were taken at 40 ns of the simulations. The aggregates of system 4 in
Discussion
Mg2+ has been poorly understood within the literature. It is known to be an inhibitor, but the clinical application remains elusive. 1 –4 Using MD, theoretical chemistry has allowed us to examine the molecular mechanism of the interaction of Mg2+ with Ca2+ and Ox2− along with other known promoters and inhibitors. This will ultimately allow for a backbone to begin clinical applications of Mg2+ in stone formation.
While theoretical modeling has been used in stone disease, there has not been a previously described application to Mg2+ particularly with the obtained calcium oxalate aggregates.
While MD is able to provide information regarding ionic interaction, there are limitations in its use. Primarily, the biggest limitation to date is the inability to run computer experiments at physiologic concentrations. While we have been able to obtain the overall ionic concentrations close to urinary conditions, the concentrations of key ions, such as calcium and oxalate, still remain significantly higher than those seen in normal human urine. Because of the time constraints of the computer simulation, these concentrations were chosen to be significantly high. In addition, these are highly controlled experiments and certainly do not contain all of the species present within human urine. As the technology advances, the experiments have included more constituents but still remain far from physiologic. Finally, this remains theoretical and the application to in vivo studies is unknown.
Further studies are warranted at this time to examine stone formation at physiologic concentrations. In addition, we would like to study Mg2+ inhibition within human models.
Conclusion
The ion pair interaction of Ca2+ and Ox2− ions is weakened in the presence of magnesium, indicating a shortened contact time. The effect of Mg2+ appears to be influenced by its density as well as its positions with respect to Ox2−. Mg2+ inhibitory effect is synergistic with citrate and remains effective in acidic environments. Further studies are needed to see if this can be applied to in vivo models as well as extending this to other stone inhibitors and promoters.
Footnotes
Acknowledgment
We are grateful to the National Institute for Computational Sciences (NICS) for the supercomputing time grant, Kraken TG-MCB100064.
Disclosure Statement
No competing financial interests exist.