
Abstract
The PulseNet Methods Development and Validation Laboratory began a re-evaluation of the standardized pulsed-field gel electrophoresis (PFGE) protocols with the goal of optimizing their overall performance and robustness. Herein, we describe a stepwise evaluation of the PulseNet-standardized PFGE protocol for Listeria monocytogenes that led to the modification of several steps which significantly improved the overall appearance and reproducibility of the resulting PFGE data. These improvements included the following: (1) reducing the cell suspension concentration, (2) increasing lysozyme incubation temperature from 37°C to 56°C, and (3) decreasing the number of units of restriction enzymes AscI and ApaI. These changes were incorporated into a proposed protocol that was evaluated by 16 PulseNet participating laboratories, including 2 international participants. Results from the validation study indicated that the updated L. monocytogenes protocol is more robust than the original PulseNet-standardized protocol established in 1998 and this resulted in the official adoption of the new protocol into the PulseNet system in the spring of 2008. The modifications not only represent an improvement to the protocol but also describe procedural improvements that could be potentially applied to the PFGE analysis of other Gram-positive organisms.
Introduction
The cell wall of Gram-positive bacteria, which is highly resistant to in situ lysis (Salazar and Asenjo, 2007), represents the most formidable challenge to consistent PFGE subtyping of such organisms. Because of this, many PFGE protocols for Gram-positive bacteria, including the PulseNet protocol for L. monocytogenes, are plagued by reproducibility and data quality problems caused by inefficient lysis. The current standardized protocol for L. monocytogenes can present difficulties when attempting to type emerging strains, because of the amount of uninterpretable data seen. The PulseNet Methods Development and Validation Laboratory began to examine the standardized PFGE protocol for L. monocytogenes to optimize and increase the efficiency of the lysis step and improve the overall protocol performance, particularly with new and hard to type strains. An additional objective of this evaluation was to adjust the format of L. monocytogenes protocol to more closely resemble that of the other standardized PulseNet PFGE protocols.
Materials and Methods
Bacterial strains
Bacteria were grown on brain heart infusion agar plates at 37°C for 14–16 hours. All isolates were identified by AccuProbe® test (GenProbe, San Diego, CA) and serotyped using the method described by Seeliger (1979). A set of 117 L. monocytogenes strains were selected for PFGE protocol development and evaluation from the Listeria Reference and Subtyping Laboratory strain collection which included surveillance and outbreak isolates that represented a variety of serotypes (Table 1).
n/a, not available.
Optimization of the PulseNet protocol for PFGE analysis of L. monocytogenes
A stepwise evaluation of the original PFGE standardized protocol for L. monocytogenes was performed and a comprehensive list of the parameters evaluated as well as the final changes incorporated into the optimized protocol are presented in Table 2. The development of an optimized PFGE protocol for L. monocytogenes was modeled after the PulseNet-standardized protocol for E. coli O157:H7 (Ribot et al., 2006) with minor modifications.
BSA, bovine serum albumin; SDS, sodium dodecyl sulfate; TE, Tris–EDTA.
Optimized L. monocytogenes protocol
Bacterial cells were removed from the plate surface using a sterile cotton or polyester fiber swab that had been moistened with Tris–EDTA (TE) buffer (10 mM Tris and 1 mM EDTA [pH 8.0], made in-house). The cells were then transferred to tubes (Falcon 2057, 12 × 75 mm; Becton Dickinson, Franklin Lakes, NJ) containing 2 mL of TE buffer. The concentration of each cell suspension was adjusted to a turbidity reading of 0.40–0.45 on a Dade Microscan Turbidity Meter (Siemens Healthcare Diagnostics, Deerfield, IL). This value corresponds to an optical density value of 0.9–1.0 measured at a wavelength of 610 nm with a spectrophotometer (Shimadzu, Kyoto, Japan) and transmittance values of approximately 17–18% when using a Vitek colorimeter (BioMérieux, Durham, NC). A 400 μL aliquot of each adjusted cell suspension was transferred to a sterile microcentrifuge tube containing 20 μL of a 20 mg/mL stock solution (in TE buffer) of lysozyme (item no. L7651; Sigma-Aldrich, St. Louis, MO). The lysozyme and cell suspension solution was manually mixed by gently tapping the capped tube against the bench top or fingers and then incubated in a 56°C water bath for 10–15 minutes. Following lysozyme incubation, a 20 μL aliquot of Proteinase K (20 mg/mL stock; Amresco, Solon, OH; Invitrogen, Carlsbad, CA) was added and mixed gently. Plugs were cast by mixing each cell suspension with 400 μL of molten 1% SeaKem Gold agarose (Lonza, Walkersville, MD) and 1% sodium dodecyl sulfate (Roche Diagnostics, Indianapolis, IN) solution that had been equilibrated for 15 minutes in a 54–56°C water bath. The mixture was immediately dispensed into two to three wells of a reusable or disposable PFGE plug mold (Bio-Rad, Hercules, CA). The resulting plugs were allowed to solidify at room temperature for 5–10 minutes and then transferred to 50-mL centrifuge tubes for subsequent lysis and plug-washing steps as described (Ribot et al., 2006). The plugs were subjected to restriction enzyme digestion or stored in TE buffer at 4°C until needed. Subsequent procedural steps, including enzyme digestions, electrophoresis conditions, and image acquisition, remained unchanged (Graves and Swaminathan, 2001), except for the reduction in the concentration of ApaI restriction endonuclease from 200 to 50 U per plug slice.
Optimized protocol validation
The procedural modifications identified during the protocol development process were incorporated into a proposed optimized protocol for PFGE analysis of L. monocytogenes. This proposed protocol was subjected to an extensive evaluation process that included internal validation by the PulseNet Methods Development and Validation Laboratory and the Listeria Reference and Subtyping Laboratory at the Centers for Disease Control and Prevention (CDC) and two external validations conducted by PulseNet participants in the public health laboratories in Connecticut, Hawaii, Los Angeles County, Massachusetts, New York City, New York State, Ohio, South Carolina, Tennessee, Virginia, and the United States Department of Agriculture Food Safety Inspection Service, Outbreak Section of Eastern Laboratory, the Food and Drug Administration (FDA) Pacific Regional Lab Northwest, the FDA PulseNet Team, and the FDA Southeast Regional Lab, the Centers for Disease Control Taiwan, the Public Health Laboratory Center Hong Kong. Each lab was asked to run and submit tiff files of two 10-well gels (5.5″ × 5″) containing both ApaI and AscI restriction patterns. The first of these gels contained four L. monocytogenes test strains that the laboratories received from CDC. For the second gel, the laboratories were requested to test four strains isolated within their region to determine the performance of the protocol with geographically diverse strains. The resulting gels were analyzed by staff from each participating laboratory as well as collectively by the PulseNet Methods and Development and Reference Laboratory at CDC. The inter- and intralaboratory reproducibility of band marking was assessed by comparing the results generated by the submitting laboratory with the analysis done at CDC. Overall image quality was also assessed by the CDC PulseNet laboratory and was determined by clarity of the restriction pattern, amount of background, and proper run length. Finally, laboratories were asked to fill out a worksheet that documented areas of potential variation between laboratories, such as equipments and vendors of various reagents.
Results
Protocol optimization
All the modifications included in the optimized PFGE protocol for L. monocytogenes are detailed in Table 2. Several procedural modifications were evaluated to determine whether the changes impacted PFGE performance. Among these were the modification of the cell suspension volume (from 300 to 400 μL), plug agarose concentration (from 1.2% to 1.0%), the addition of Proteinase K to the cell suspension rather than the plug agarose, the volume of Proteinase K added to the cell suspension (from 3 to 20 μL), and the volume of lysis buffer used (from 4 to 5 mL). Testing of these modifications demonstrated no significant difference in the resulting PFGE patterns (data not shown), but their inclusion in the proposed protocol made the PFGE process for L. monocytogenes more consistent with PulseNet-standardized PFGE protocols for other bacteria (Ribot et al., 2006).
Additional testing was performed to identify areas for optimization to enhance overall PFGE protocol performance. Cell suspension concentration values were tested over the range of 0.2–0.8, as measured by a Dade microscan turbidity meter. A range of 0.4–0.45 was selected because it consistently produced DNA fragments of even intensity throughout the L. monocytogenes PFGE pattern, while resulting in minimal background compared with higher cell suspension concentrations (data not shown).
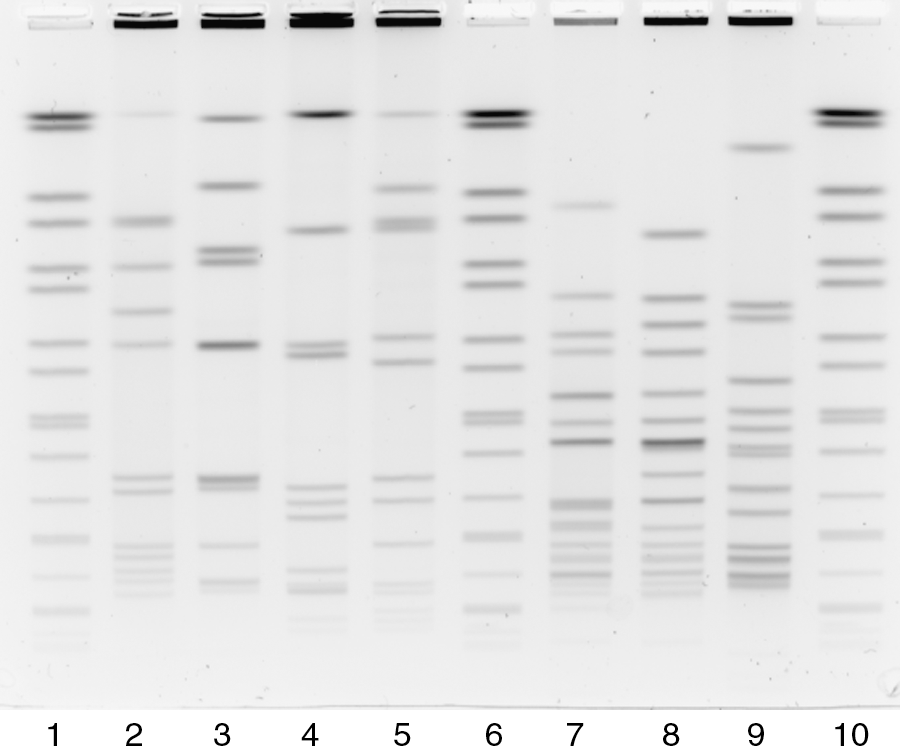
The lysozyme treatment of L. monocytogenes cells was thoroughly evaluated and several areas for optimization were identified. For example, the lysozyme diluent was changed from water to TE buffer because of increased solubility within TE buffer (20 mg/mL in TE buffer vs. 10 mg/mL in water) (data not shown). Treatment of the cell suspensions with lysozyme was evaluated with respect to final concentration (0.5–2.5 mg/mL), incubation time (0, 5, 10, 15, 20, and 30 minutes), and incubation temperature (37°C and 56°C). Increasing the lysozyme incubation temperature to 56°C greatly increased the efficiency of cell lysis (Fig. 1). At this temperature, incubating the cell suspension with a final concentration of 0.49 mg/mL lysozyme for just 10 minutes proved sufficient for complete lysis of L. monocytogenes. Following the application of these changes, the amount of restriction enzymes, ApaI and AscI, sufficient for complete digestion of the plug slice within 2 hours was reduced from 200 to 50 U (data not shown). Figure 3 shows the Listeria PulseNet certification strains after incorporating all of the aforementioned changes.

Lanes 1, 7, 13: H9812 PulseNet global standard digested with XbaI. Lanes 2–6: tested at 37°C with increasing lysozyme incubation times (5, 10, 15, 20, and 30 minutes) and digested with ApaI. Lanes 8–13: tested at 56°C with increasing lysozyme incubation times (5, 10, 15, 20, and 30 minutes) and digested with ApaI.
Protocol validation
To assess whether this protocol was robust and yielded reproducible results across multiple laboratories, an extensive external validation was undertaken. Figure 2 shows a representative PFGE gel containing the four validation isolates digested with either AscI (lanes 2–5) or ApaI (lanes 7–9) tested by each of the laboratories. Assessment of overall gel quality revealed that 81% of the laboratories involved in the validation submitted gels of good to excellent quality. Comparison of the PFGE patterns from the validation strains tested revealed greater than 90% similarity between the submitting laboratory's analysis and that done by the PulseNet Methods and Development Laboratory, demonstrating a high degree of reproducibility (BioNumerics parameters: dice coefficient, unweighted pair group method with arithmetic mean (UPGMA), optimization, and tolerance set at 1.50) (data not shown). Also, each laboratory submitted PFGE data on L. monocytogenes isolates received within their geographic region. As with the validation strains, the majority of these gels were of good to excellent quality and in several instances resulted in the generation of a PFGE pattern for isolates the laboratory was previously unable to type with the original PulseNet protocol.
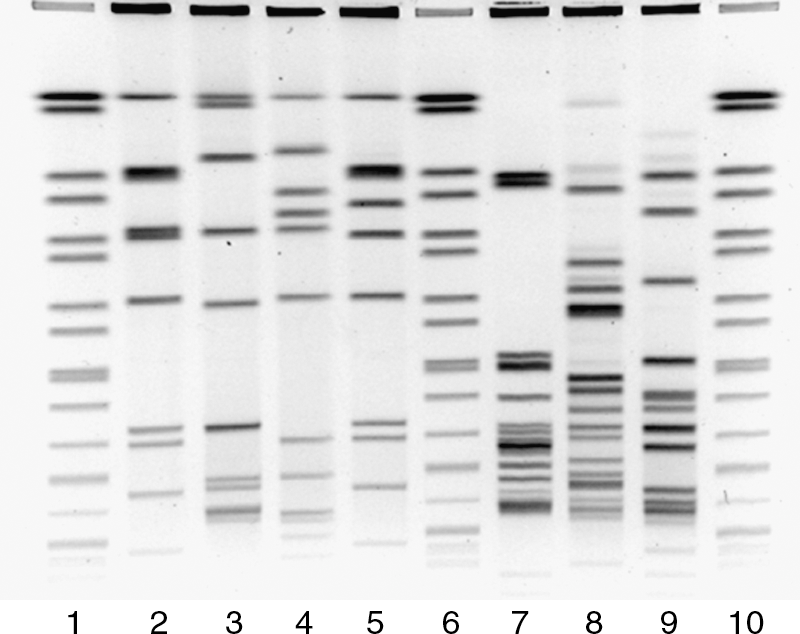
Example of a Listeria monocytogenes gel submitted by a public health laboratory during the external validation phase.
Discussion
An efficient PFGE protocol depends on achieving a balance among multiple variables, including cell suspension concentration, lysis efficiency, and DNA endonuclease restriction. A reevaluation of procedural steps for PFGE analysis of L. monocytogenes was undertaken with the goal of increasing the overall efficiency of the protocol. Additionally, it was hoped that a corresponding decrease in time to obtain results as well as costs would accompany the improvement in protocol performance. Various conditions were evaluated to determine any of the parameters that could be optimized in the previously established PulseNet-standardized protocol for L. monocytogenes. Special attention was given to the cell suspensions, plug preparation, and DNA endonuclease restriction. Multiple parameters were evaluated and the most appropriate was implemented to make the protocol more consistent in structure with other PulseNet-standardized PFGE procedures (e.g., E. coli O157), in an effort to reduce reagent and procedural differences between the protocols. A summary of the modifications is included in Table 2.
In PFGE analysis, achieving proper DNA fragment (band) intensity depends on establishing a balance between cell suspension concentration, efficient lysis, and concentration of released DNA to be cleaved in the desired period of time. Identification of an optimal cell suspension concentration was demonstrated to be an important factor in the development of the standardized PulseNet protocol for V. cholerae (Cooper et al., 2006) because of the fact that higher DNA concentration have the potential to yield thicker, less distinct bands that can complicate pattern analysis because of poor resolution of closely migrating fragments. The potential impact of cell suspension concentrations on lysis efficiency was also considered for Listeria. If cell suspension concentrations are below an appropriate threshold, banding patterns appear faint, particularly among smaller DNA fragments that retain less ethidium bromide because of a low concentration of DNA. In contrast, cell suspension concentrations that are too high lead to incomplete lysis and subsequent incomplete DNA restriction because in part of an increase in residual cellular and chemical impurities that can be difficult to remove completely during washing steps. This situation can also yield faint patterns, which may lead the investigator to increase the cell suspension concentration in an effort to compensate for the low level of available DNA. Increasing the cell suspension concentration when poor lysis is observed will, however, disrupt the balance of bacterial cells and chemical components involved in cell lysis and not necessarily produce better results. We recognized that it was important to identify an appropriate cell suspension concentration to achieve that balance and ensure optimal levels of lysis. After testing a wide range of cell suspension concentrations, a turbidity range of 0.40–0.45 on a Dade Microscan turbidity meter was selected because it consistently produces DNA fragments of even intensity throughout L. monocytogenes PFGE patterns with little or no background (data not shown).
One of the main issues observed with L. monocytogenes PFGE is the presence of faint patterns with high residual DNA fluorescence observed within the plug slice of the well. These results are typically associated with incomplete cell lysis, a common problem with PFGE analysis of Gram-positive bacteria. Lysis of Gram-positive bacteria occurs after enzymatic treatment with lysozyme and/or other proteolytic enzymes (Salazar and Asenjo, 2007) that disrupt the cell wall and allow access to the DNA. Consequently, the efforts to optimize this protocol focused on enhancing cell lysis efficiency in the early steps of the PFGE process. Traditionally, lysozyme treatment of cell suspension concentrations occurs at 37°C, but the enzyme remains active until denaturation around 65°C (Masumoto et al., 2000). Despite an exhaustive literature search, we did not identify studies that established the optimal incubation temperature for lysozyme in the context of PFGE or lysis of Gram-positive bacteria, and therefore, the effect of incubation temperature on lysozyme treatment of cell suspension concentrations was evaluated. Increasing the lysozyme incubation temperature from 37°C to 56°C markedly improved cell lysis efficiency (Fig. 1). Once this variable was identified, factors such as lysozyme concentration and incubation time were tested to identify the most appropriate values. A higher stock concentration (20 mg/mL in TE vs. 10 mg/mL in water) was made to increase final lysozyme concentration in the plug mixture without increasing final volume. The lysozyme solution appears to be more stable in TE buffer than water, thereby reducing time and expense associated with routine preparation and handling of this reagent, as well as limiting lysozyme degradation which decreases lysis efficiency and results in subsequent assay failure.
Increased cell lysis efficiency leads to an increase in the amount of accessible DNA, even with lower cell suspension concentration. Reduced cell suspension concentrations also result in a reduction of chemical and cellular components that may inhibit DNA digestion, and therefore, fewer units of restriction endonuclease are necessary to achieve complete DNA digestion. Following the optimization of the plug preparation procedure, the amount of enzyme necessary for DNA digestion was investigated. Our results demonstrated that 50 U of both ApaI and AscI is sufficient for complete digestion of the plug slice in 2 hours (data not shown). This represents a 75% reduction in the number of ApaI units required per reaction compared with the original PulseNet protocol and an equivalent cost reduction per reaction. Figure 3 shows a gel that incorporates all of these changes, using the PulseNet Listeria certification set. There was significant improvement in lysis and complete digestion with the reduced ApaI units and reduced incubation time.
PulseNet L. monocytogenes certification strains prepared with the optimized protocol. Lanes 1, 6, 10: H9812 PulseNet global standard digested with XbaI. Lanes 2–5: H8393, H8394, H8395, and H2446 digested with AscI. Lanes 7–10: H8393, H8394, and H8395 digested with ApaI.
Overall, the data submitted during the external validation process were of good to excellent quality and demonstrated a relatively high degree of reproducibility. In addition to testing known strains, participating laboratories were asked to perform PFGE analysis using the proposed protocol on L. monocytogenes strains isolated within their own state or region. Interestingly, several laboratories reported the ability to type isolates that they previously had difficulty typing or for which they were unable to obtain a pattern by using the original standardized PulseNet protocol. In each of these situations, the laboratories were able to generate a pattern with the proposed protocol that, when compared with the pattern generated previously, demonstrated a marked improvement in overall pattern appearance (data not shown).
Real-time foodborne disease surveillance depends on the ability to type the majority of isolates in a rapid manner. Because of the public health importance of L. monocytogenes, a 3-day turnaround time is expected for the submission of subtyping data from PulseNet participating laboratories. Repeat testing or the need to submit these isolates to a reference laboratory for subtyping may delay L. monocytogenes cluster detection. These results demonstrated that this protocol is more robust, may reduce the failure rate observed by participating PulseNet laboratories, and increase the speed at which data are submitted to the national databases and in turn the detection of possible outbreaks.
Conclusions
Over the last several decades, PFGE has been firmly established as the gold standard subtyping method for a large number of bacterial species. Widespread use of this technique has resulted in a greater understanding of the procedure, and continual efforts to improve and optimize protocols have been made. The improved lysis and associated procedural modifications described here represent a marked improvement in quality, turnaround time, and cost of the PFGE protocol for L. monocytogenes and these have been adopted into the standardized PulseNet PFGE protocol for L. monocytogenes in the spring of 2008. Traditionally, Gram-positive bacteria have presented significant problems in producing reproducible and high-quality data using PFGE. Although these modifications were specifically developed for L. monocytogenes, it is likely that the current improvements may be applicable to PFGE analysis of other Gram-positive organisms.
Footnotes
Acknowledgments
The authors acknowledge the support from the Connecticut Department of Public Health Laboratory, the Hawaii State Department of Health, the Los Angeles County Public Health Lab, the Massachusetts Department of Public Health, the New York City Department of Health and Mental Hygiene, the New York State Department of Health, the Ohio Department of Health, South Carolina Department of Health and Environmental Control, Tennessee Department of Health Laboratory Services, Virginia Division of Consolidated Laboratories, the United States Department of Agriculture/Food Safety Inspection Service/Outbreak Section of Eastern Laboratory, the FDA Pacific Regional Lab NW, the FDA PulseNet Team, the FDA Southeast Regional Lab, the Centers for Disease Control Taiwan, and the Public Health Laboratory Center (Hong Kong). Jessica L. Halpin was supported by a fellowship from the Oak Ridge Institute for Science and Education.
Disclosure Statement
No competing financial interests exist.