
Abstract
To achieve an effective detection of Salmonella spp., Escherichia coli O157, and Listeria monocytogenes in meat products, a multiplex real-time polymerase chain reaction (PCR) coupled with a multipathogen enrichment strategy was developed in this study. Pathogen-specific DNA sequences in the invA, rfbE, and hlyA genes were employed to design primers and TaqMan probes for identifying Salmonella spp., E. coli O157, and L. monocytogenes, respectively. An internal amplification control (IAC) utilizing a novel DNA sequence from human adenovirus was incorporated into the multiplex PCR assay to indicate false-negative results. Concurrent amplifications of multiple targets and IAC were thoroughly evaluated and optimized to minimize PCR competitions. Combined with a multipathogen enrichment in a selective enrichment broth for Salmonella, Escherichia, and Listeria (SEL), the multiplex real-time PCR assay was able to simultaneously detect all of the three organisms in artificially contaminated ground beef at a detection sensitivity of <18 CFU/10 g ground beef. Applying the assay to 26 retail meat samples including beef, chicken, turkey, and pork revealed that 12 samples were positive for one of the organisms and 3 samples were positive for two of the organisms after a 20-h enrichment in SEL. The remaining meat samples tested negative for all of the organisms by only showing amplification of the IAC. These results were confirmed by traditional culture methods testing for each individual species. Taken together, the multiplex real-time PCR assay combined with multipathogen enrichment is a rapid and reliable method for effectively screening single or multiple pathogen occurrences in various meat products.
Introduction
Common sources for transmitting these foodborne pathogens are raw meat including beef, pork, and poultry products. Other food products such as milk, cheese, eggs, vegetables, and ready-to-eat food also could be contaminated with these pathogens. Consumption of food contaminated by these pathogens has not only caused numerous infections but also resulted in numerous foodborne outbreaks. For example, a recent outbreak associated with peanut butter contaminated with Salmonella serotype Typhimurium infected 529 people in 43 states (CDC, 2006). Such facts have caused concerns about the safety of food to both industry representatives as well as to regulatory agencies (Schuchat et al., 1991; Rose et al., 2002; Uhlich et al., 2008).
To better control microbial contaminations of food and consequently to reduce foodborne illnesses, rapid and accurate pathogen detection methods are required for effectively monitoring microbial pathogens in food supplies. Traditional detection methods depend upon selective plating combined with immunological or biochemical identification. The negative aspects of these methods are that they are time consuming, laborious, and take several days to complete. In fact, it is impractical to use traditional microbiological methods for high-throughput screening of large numbers of food samples for the presence of one or more pathogens (Abubakar et al., 2007).
Real-time polymerase chain reaction (PCR) is a promising molecular tool for identifying microorganisms (Espy et al., 2006). It not only has the sensitivity and specificity of traditional PCR (i.e., can detect a few copies of target genomic DNA) but also can display the results during amplification. Real-time PCR does not require DNA electrophoresis and associated visualization with ethidium bromide staining. Moreover, TaqMan-based real-time PCR methods allow multiple sequence amplifications to be detected in a single reaction with labeled probes, which have spectrally distinct fluorescent properties. Thus, for pathogen detection, several species can be identified simultaneously based upon use of specific primers and probes with corresponding fluorescent signals.
There have been few reports of multiplex real-time PCR detection of Salmonella spp., E. coli O157:H7, and L. monocytogenes in food (Bhagwat, 2003; Nguyen et al., 2004; Wang et al., 2007; Elizaquivel and Aznar, 2008). However, none of these multiplex assays included an internal amplification control (IAC) except for a singleplex detection of Salmonella enterica or L. monocytogenes in food (Murphy et al., 2007). IAC is a nontarget DNA sequence coamplified with target DNA in the same PCR tube and is used to discriminate a false-negative result from a true-negative result in PCR detection. As PCR failure could be due to inhibitory food residues, organic solvents from nucleic acid extraction, or other errors, IAC has been considered to be necessary in PCR analysis of food samples (Burggraf and Olgemoller, 2004).
The aim of this study was to develop a reliable and effective multiplex real-time PCR assay to simultaneously detect Salmonella spp., E. coli O157, and L. monocytogenes in food. A novel IAC that shares no sequence homology with any bacterial genomes was designed and optimized in the multiplex PCR for the indication of false-negative results. To provide sufficient numbers of target pathogens for detection, a multipathogen enrichment strategy was employed to concurrently enrich these three pathogens prior to multiplex real-time PCR detection (Kim and Bhunia, 2008). Finally, the application of the method was extensively evaluated in both artificially and naturally contaminated meat samples.
Materials and Methods
Bacterial strains and growth conditions
The bacterial reference strains used in this study are listed in Table 1. The Campylobacter strains were grown at 42°C in Brucella medium (Becton Dickinson, Sparks, MD) in a microaerobic workstation (Don Whiteley Scientific, Shipley, UK) with 85% N2, 10% CO2, 5% O2, and 82% relative humidity. The rest of the strains were aerobically grown at 37°C in Brain Heart Infusion (BHI) medium (Becton Dickinson) with shaking at 150 rpm except for Carnobacterium mobile and Brochothrix thermosphacta, which were grown at 30°C in BHI medium without shaking.
IAC, internal amplification control; PCR, polymerase chain reaction.
Internal amplification control
Based on the sequence exclusivity to bacterial genomes, a 79-bp DNA sequence of the human adenovirus (accession no. AY601634) was selected as an IAC for the multiplex PCR assay. The IAC DNA fragment was PCR amplified from a long oligonuleotide (TGGAAGCAATGCCAAATGTGTATGTGGTGGCATTGTCTTCTCCCGTTGTAACTATCCACTGAGATGTGTTAGGCGCGCC) using IAC-F/R primers (Table 2). After purification with a QIAquick gel extraction kit (Qiagen, Valencia, CA), the 79-bp amplicon was quantified and used as the IAC in the multiplex real-time PCR assay.
Primer and probe design
The DNA sequences of the invA, rfbE, and hlyA genes from the targeted microorganisms were retrieved from GenBank (
Genomic DNA extraction
One milliliter of freshly grown bacterial culture was centrifuged at 8000 g for 10 min. The cell pellets were subjected to DNA extraction using the DNeasy Blood and Tissue kit (Qiagen), following the manufacturer's recommendations. For Gram-positive bacteria, cells were digested in 180 μL of 20 mg/mL lysozyme (containing 20 mM Tris-Cl, pH 8.0, 2 mM sodium ethylenediaminetetraacetic acid, and 1.2% Triton® X-100) at 37°C for 30 min, and then the Qiagen kit was used to extract DNA. The genomic DNA extraction of pathogens from food samples was the same procedure as used for extraction of DNA from Gram-positive bacteria. After purification, DNA was eluted with 100 μL ddH2O. The DNA quantity (A 260) and quality (ratio of A 260/A 280) were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE).
Multiplex real-time PCR
Multiplex real-time PCR was performed on a 7500 Real-Time PCR system (Applied Biosystems). Each 20 μL reaction mixture contained 1 × TaqMan® Gene Expression Master Mix (Applied Biosystems), four sets of primers and probes with a concentration of 200 nM each, 1.2 × 104 copies of IAC, and 2 μL sample DNA. Real-time PCR was performed in triplicate with the following program: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min. To optimize the IAC concentration, 10-fold serial dilutions of the IAC ranging from 101 to 106 copies/reaction were mixed with a DNA mixture containing 102 copies per reaction of each genomic DNA from Salmonella enterica Enteritidis ATCC 13076, E. coli O157:H7 EDL933, and L. monocytogenes ATCC 19111. PCR amplification efficiency and detection sensitivity were determined using a series of 10-fold dilutions of genomic DNA from a single strain or a mixture of genomic DNA from the three strains. The amplification efficiencies were calculated from the following formula: E = (10−1/slope) − 1, using the slopes of the linear standard curves by plotting cycle thresholds against the log10 of genomic copies of each DNA per reaction.
Examination of artificially contaminated ground beef
Inoculated from overnight cultures, the freshly grown cells of Salmonella Enteritidis ATCC 13076, E. coli O157:H7 EDL933, and L. monocytogenes ATCC 19111 in the late exponential phase (approximately 108 CFU/mL) were used to spike 86% lean ground beef (Wegmans, Warrington, PA). The ground beef was irradiated before purchasing and confirmed to be lacking in the three target pathogens before spiking. The procedures in the U.S. Department of Agriculture/Food Safety and Inspection Service (FSIS) Microbiology Laboratory Guidebook were used for preparing meat samples with the following modifications. Each sample was analyzed in duplicate and the experiment was independently repeated three times. Briefly, 1 mL of 10-fold serial dilutions of the bacterial cells from 0 to 104 CFU/mL was inoculated into 10 g of ground beef in a filter stomacher bag (Nasco Whirl-Pak, Fort Atkinson, WI). Subsequently, 90 mL of buffered Listeria enrichment broth base (Becton Dickinson) was added into each bag, and the samples were homogenized for 2 min using a Stomacher 400 (Seward, Norfolk, UK). The homogenized samples were incubated at 37°C for 1 h with shaking at 150 rpm before adding selective antibiotics to achieve final concentrations of 0.01 g/L for acriflavine (ICN Biomedical, Aurora, OH), 0.05 g/L for cycloheximide, 0.05 g/L for fosfomycin, and 0.002 g/L for nalidixic acid (Sigma, St. Louis, MO) as described for SEL, a multipathogen selective enrichment broth (Kim and Bhunia, 2008). After 6- or 20-h incubation times, 1 mL aliquot was collected from each sample for DNA extraction. In parallel, the same sample was plated for viable cell counts (CFU) on the selective agars: CHROMagar Salmonella for Salmonella spp., CHROMagar O157 for E. coli O157:H7, and CHROMagar Listeria for L. monocytogenes (Becton Dickinson).
Examination of naturally contaminated meat
Twenty-six independent meat samples including beef, chicken, turkey, and pork were obtained from local supermarkets, kept at 4°C, and tested within 12 h after purchasing. Each of the meat samples was analyzed in duplicate. For testing, 25 g of meat was placed in a filter bag containing 225 mL of buffered Listeria enrichment broth base. The homogenization and selective enrichment of the pathogens in these naturally contaminated samples were carried out with the same procedure as the spiked samples. After a 20-h enrichment, the samples were subjected to DNA extraction and multiplex real-time PCR analysis and culture-based detection for the three pathogens in parallel. The enrichments were plated onto CHROMagar O157 and sorbitol MacConkey agar with cefixime–tellurite (CT-SMAC agar) for E. coli O157:H7, CHROMagar Salmonella and xyloselysine–deoxycholate agar for Salmonella spp., and modified Oxford agar for L. monocytogenes. The E. coli O157:H7 serotype was further assessed by a latex test using RIMTM E. coli O157:H7 (Remel, Lenexa, KS). Listeria was verified by API Listeria strips (Biomerieux, Durham, NC).
Results
Design and evaluation of an IAC
To develop a reliable PCR method for foodborne pathogen detection, an IAC was designed and included in the multiplex real-time PCR to indicate false-negative results. The assay is comprised of four sets of primers and TaqMan probes for the targets invA, rfbE, hlyA, and IAC. In testing 76 different bacterial strains, the IAC primers and probes did not show any amplification from the genomic DNA of these strains, suggesting a high exclusivity of the IAC sequence to bacterial DNA (Table 1). As the targets and the IAC are coamplified in the same tube, it becomes necessary to titrate the amount of IAC to be added so that the interference between the targets and the IAC would be minimized. To optimize the concentration of the IAC used in the multiplex PCR, a series of 10-fold IAC dilutions ranging from 106 to 101 DNA copies was coamplified with a mixed DNA solution that contained approximately 102 copies of each genomic DNA from Salmonella spp., E. coli O157:H7, and L. monocytogenes. As a noncompetition control, the same IAC dilution series was amplified in a singleplex real-time PCR in the presence of the IAC primers and probes only. The results are shown in Figure 1. The Ct of the IAC from both singleplex and multiplex PCR displayed similarly increased values as the IAC concentrations were decreased from 106 to 104 copies/reaction, indicating no inhibition of the IAC amplification in the multiplex PCR when the IAC was used at 104 copies/reaction or higher. However, when the IAC concentration was lower than 104 copies/reaction, the Ct of the IAC from the multiplex amplification was either higher than that from the singleplex PCR or undetectable (>40), suggesting some inhibition on IAC amplification at the low concentrations. Of the target amplifications that occurred in the same reaction tube, the Ct values of invA and hlyA remained the same regardless of the 106 to 100 copies of the IAC, thus suggesting that the presence of the IAC had no effect on invA and hlyA amplifications. However, the amplification of rfbE was slightly inhibited by the IAC at 105 copies or higher. Therefore, 104 copies/reaction of the IAC was determined as the optimal concentration for the multiplex real-time PCR assay.
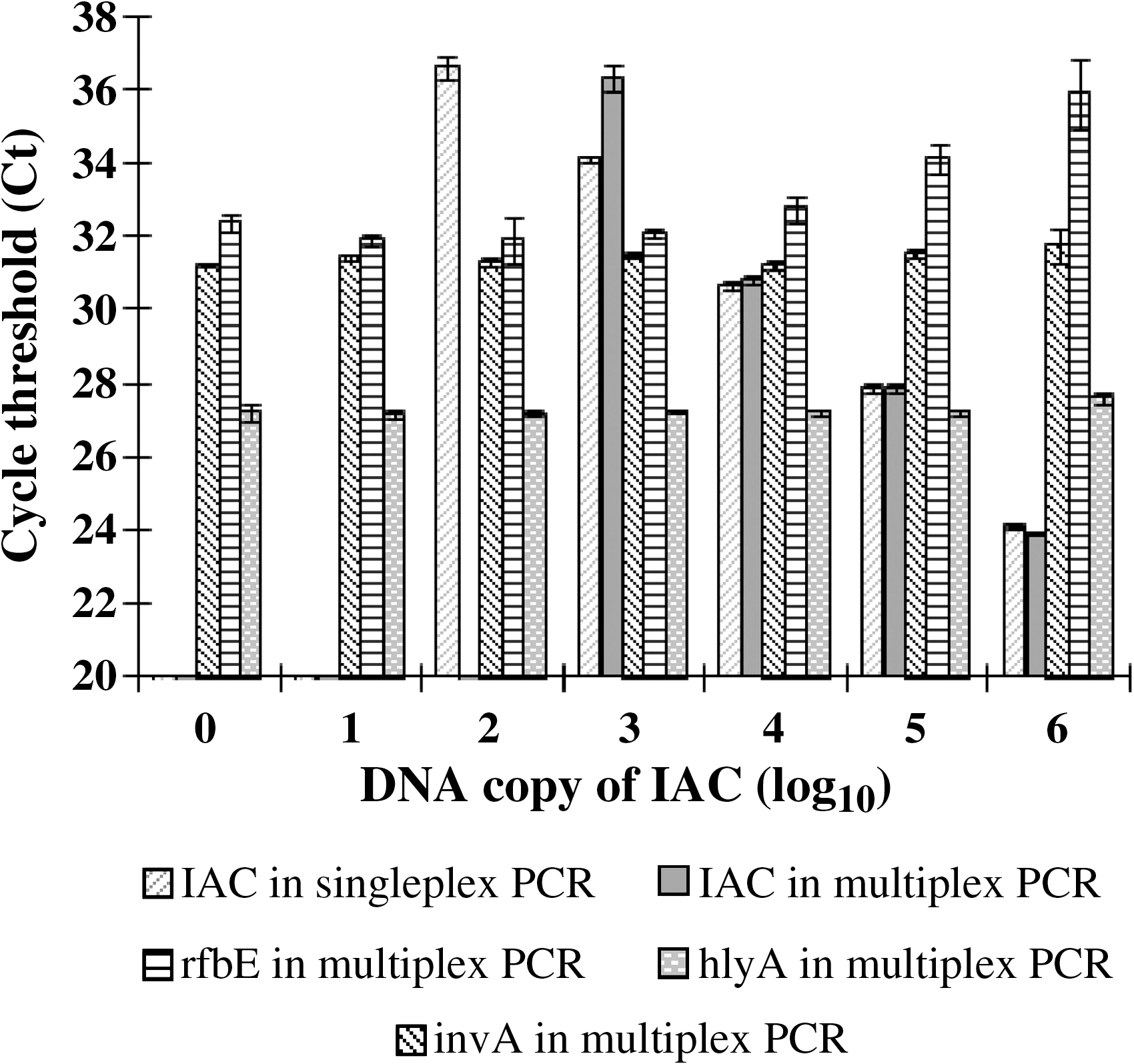
The effect of internal amplification control (IAC) concentration on multiplex real-time polymerase chain reaction (PCR) amplification. In the multiplex real-time PCR containing four sets of primers and probes for invA, rfbE, hlyA, and the IAC, approximately 102 copies of each genomic DNA from Salmonella Enteritidis ATCC 13076, Escherichia coli O157:H7 EDL933, and Listeria monocytogenes ATCC 19111 were coamplified with a series of 10-fold dilutions of the IAC. In the singleplex real-time PCR control, only the IAC template and a set of primers and probes for IAC were presented.
When a fixed concentration (104 copies/reaction) of the IAC was used together with a genomic DNA mixture of Salmonella spp., E. coli O157:H7, and L. monocytogenes (Table 3) in the range of about 107 to 101 genome copies in total, the IAC amplification was not inhibited until the genomic DNA reached at 1.5 × 104 copies per PCR reaction or higher. At such high concentrations, target DNA is easily detectable. In this case, the amplification of the IAC was unnecessary as PCR inhibition usually occurred when neither the IAC nor the target DNA was amplified. So the constrained amplification of the IAC in the presence of excess genomic DNA should not affect IAC functioning as a false-negative indicator. Similar inhibition of the IAC amplification was observed by other investigators and was considered to have no effect on the function of the IAC as well (Hoorfar et al., 2004).
The undiluted genomic DNA mixture (1.57 × 107 copies/reaction) contains 8.20 × 106 copies of DNA from Salmonella Enteritidis ATCC 13076, 5.36 × 106 copies of DNA from E. coli O157:H7 EDL933, and 2.14 × 106 copies of DNA from L. monocytogenes ATCC 19111 in each reaction.
The IAC was used at 1.20 × 104 copies/reaction.
The Ct was averaged from three independent experiments, with two replicates in each PCR run.
Ct was undetectable in 40 cycles of PCR.
Performance of the multiplex real-time PCR
To determine the efficiency in amplifying the targets of Salmonella spp., E. coli O157:H7, and L. monocytogenes, the standard curves for the multiplex PCR assays were generated with template DNA from a single pathogen or with a mixture of DNA from the three pathogens (Fig. 2). The amplification efficiencies (E) were calculated from the slopes (−3.248 to −3.572) of the linear standard curves using the following formula: E = (10−1/slope) − 1.
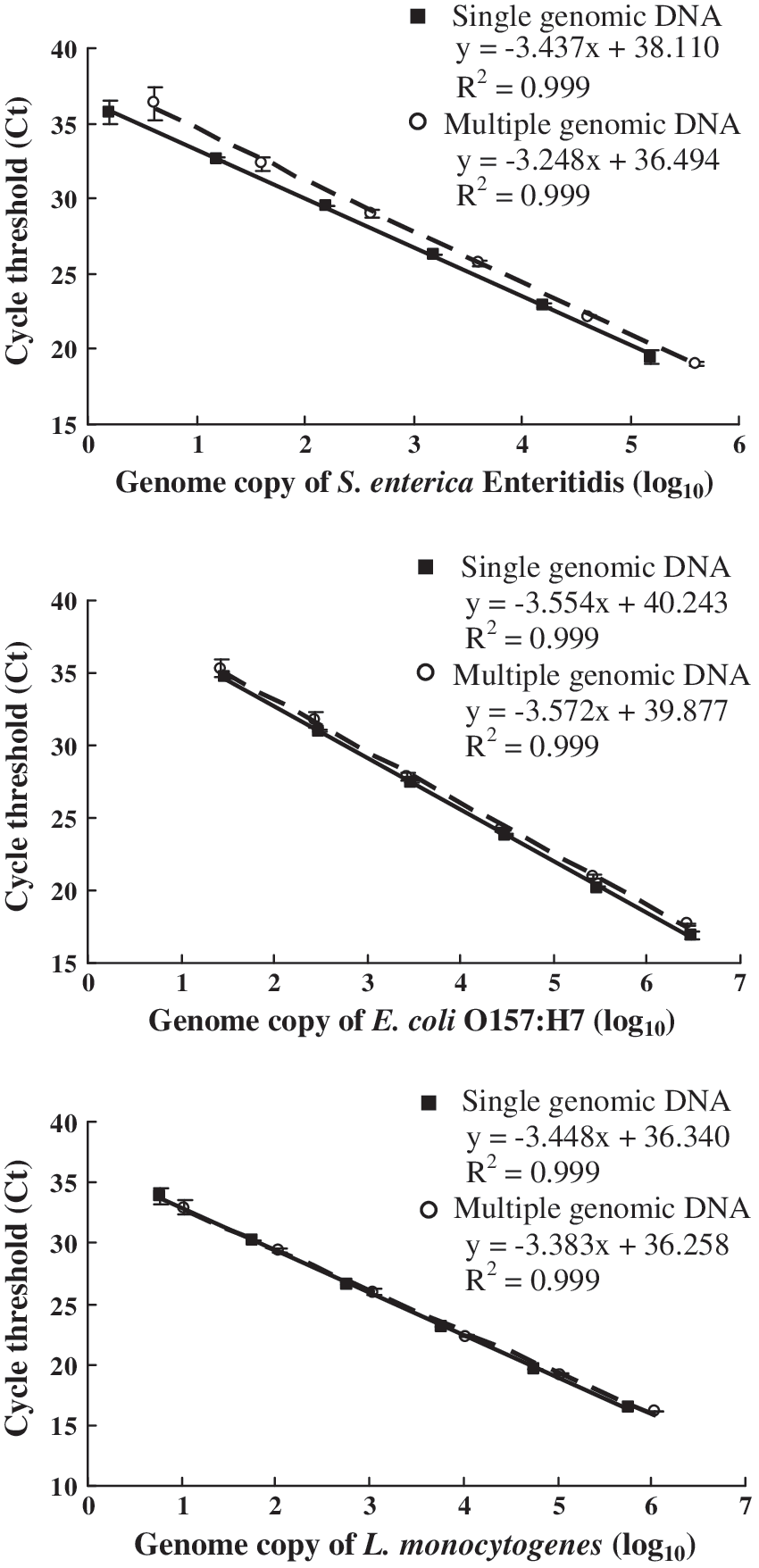
Standard curves of the multiplex real-time PCR analysis. The multiplex real-time PCR was performed for simultaneous detection of the invA, rfbE, hlyA targets and the IAC in the presence of single genomic DNA or multiple genomic DNAs from Salmonella Enteritidis ATCC 13076, E. coli O157:H7 EDL933, and L. monocytogenes ATCC 19111, as well as the IAC template. The mean Ct values of three replicates are plotted against the corresponding genome copies of DNA in log10. The solid line represents that a single genomic DNA was used in each reaction and the dotted line represents that a mixed genomic DNA from the three strains was used in each reaction.
In the multiplex real-time PCR, the amplification efficiencies were greater than 90% for all three coexisting targets, which were highly comparable to those using single genomic DNA in the same system. These results suggested that there was no noticeable competition in coamplifying the three targets in the same tube. In addition to the high amplification efficiency, the assay had a linear range of quantification over 6 orders of magnitude (from 1.57 × 101 to 1.57 × 107 copies of the mixed genomic DNA per reaction) and a detection limit of eight genome copies per reaction or lower for those three organisms (Table 3). The detection limit was deduced from the lowest concentration (1.57 × 101 copies/reaction) of the detected genomic DNA mixture, which contains approximately eight, five, and two copies of genomic DNA from Salmonella Enteritidis ATCC 13076, E. coli O157:H7 EDL933, and L. monocytogenes ATCC 19111, respectively.
To examine the specificity of the primers and probes experimentally, a total of 76 reference bacterial strains including 17 Salmonella spp., 18 E. coli O157, 5 E. coli non-O157, 15 L. monocytogenes, 6 Listeria non-monocytogenes, and 15 other strains were tested. Approximately 106 copies of each genomic DNA were assessed for reactivity in the multiplex real-time PCR system containing four sets of primers and probes targeting Salmonella spp., E. coli O157 serotype, L. monocytogenes, and the IAC. To consistently collect and analyze data, the threshold of PCR amplification profiles was set to 0.1 for all the targets (invA, rfbE, hlyA, and IAC). The results are summarized in Table 1. All the Salmonella spp., E. coli O157, and L. monocytogenes strains tested were exclusively positive for the invA, rfbE, and hlyA targets, respectively, with Ct values between 16 and 19. The rest of the strains were negative for all these targets, with undetectable Ct in 40 cycles of PCR. These results demonstrated that the assay was highly specific to Salmonella spp., the E. coli O157 serotype, and L. monocytogenes, and there was no cross-reactivity to the E. coli non-O157 serotype, other species of Listeria, or other bacteria.
Multiplex real-time PCR detection of artificially contaminated ground beef
To examine the ability of the multiplex real-time PCR assay to simultaneously detect the three pathogens in food matrices, artificially contaminated ground beef was used for the experiment. A series of 10-fold diluted culture mixtures of S. enterica Enteritidis ATCC 13076, E. coli O157:H7 ATCC 43895, and L. monocytogenes ATCC 19111 was spiked into 10 g irradiated ground beef. After a 6- or 20-h enrichment in SEL, the samples in duplicate were analyzed using the multiplex real-time PCR and verified by determining viable cell counts on selective CHROMagar plates in parallel. The results shown in Table 4 indicate, after a 6-h enrichment, all of the three pathogens were detected at a sensitivity of 18 CFU/10 g of ground beef or even lower via both methods. Under the same assay conditions, the unspiked ground beef was negative for all these targets except the IAC, indicating that the irradiated ground beef indeed did not contain any of these pathogens. As expected, the samples after a 20-h enrichment had substantially higher viable cell numbers and genome copies for all three species than the samples enriched for 6 h. Consequently, the longer enrichment (20 h) has allowed both assays to detect samples inoculated with lower numbers (2 CFU/10 g) of target pathogens. Thus, the multiplex real-time PCR was able to sensitively detect all three pathogens simultaneously in the spiked ground beef after a 6- or 20-h enrichment in SEL.
The viable cells were counted on selective CHROMagar plates after the enrichment.
The genome copies were calculated from the standard curves in Figure 2.
In SEL, E. coli O157:H7 seemed to grow faster and reach higher cell numbers than Salmonella and L. monocytogenes after a 6- or 20-h enrichment, suggesting SEL may promote E. coli O157:H7 growth better than the other two. L. monocytogenes displayed the slowest growth, having a starting concentration of 2 CFU/10 g ground beef and a final cell count of 105 CFU/mL after a 20-h coenrichment of the three species in SEL. Clearly, this slow rate of growth is not an issue because the final cell count is considerably higher than the detection limit of the multiplex real-time PCR assay.
When 1.2 × 104 copies/reaction of the IAC was used in the multiplex PCR assay in meat samples, some inhibition of amplification of the IAC was observed when the samples contained 7.4 × 105 CFU/mL (equivalent to 1.5 × 104 genome copies/reaction) or higher of these pathogens (Table 4). This is consistent with the data from pure bacteria cultures in Table 3, suggesting good quality of DNA was obtained from these pathogens in meat by the DNeasy Blood and Tissue kit.
Multiplex real-time PCR detection of naturally contaminated meat
To further validate the applications of the multiplex real-time PCR assay in food, 26 fresh meat samples including beef, chicken, turkey and pork were purchased from local supermarkets and tested for the occurrences of Salmonella, E. coli O157, and L. monocytogenes. After a 20-h enrichment in SEL, a total of 15 samples (57.7%) were found to be positive for one or two of these organisms by the multiplex real-time PCR assay. The rest of the meat samples were shown to be negative for all of these species by the positive amplification of IAC occurring in the same reaction tubes (Table 5). In the contaminated samples, eight (30.8%) were positive for Salmonella spp., three (11.5%) were positive for E. coli O157 serotype, and seven (26.9%) were positive for L. monocytogenes. Interestingly, one ground beef and one ground pork sample were found to be contaminated by both Salmonella spp. and E. coli O157, and one ground chicken sample was found to be contaminated by both Salmonella spp. and L. monocytogenes, which suggests there might be multiple pathogen occurrences in these meat samples.
To verify the multiplex real-time PCR results, the same samples were examined for each of these pathogens, using the culture-based methods described in the Materials and Methods section. The results were in agreement with the multiplex real-time PCR data, except that one ground chicken sample was found to be positive for L. monocytogenes by the multiplex real-time PCR assay but was negative by the culture- and immuno-based methods (Table 5). This discrepancy could be due to some potential PCR contamination with genomic DNA from the pathogens.
To find out if E. coli O157:H7 was presented in the three meat samples positive for rfbE, CHROMagar O157 followed by CT-SMAC agar was used to isolate the pathogen. The sorbitol-negative colonies, indicative of typical E. coli O157:H7, were isolated from all three meat samples. Agglutination assays confirmed that the isolates from both ground pork packages were E. coli O157:H7. The isolates from the ground beef package showed weak agglutinations to O157 and H7 antibodies (data not shown), which was likely due to insufficient expression of the antigens under the assay conditions (Narang et al., 2009).
Overall, the results from testing naturally contaminated meat samples further proved the feasibility of the multiplex real-time PCR assay combined with multipathogen enrichment in detecting these pathogens in meat products. The effectiveness of the assay allows it to simultaneously screen various food samples for multiple pathogen contamination.
Discussion
In this study, a multiplex real-time PCR assay was developed that used four sets of newly designed primers and probes labeled with a unique combination of fluorescent reporters to simultaneously detect Salmonella spp., E. coli O157, and L. monocytogenes in various meat samples. The major improvements of the current method from the previously reported methods are (1) the inclusion of a novel IAC to rigorously control false-negative results and (2) the combination of multiple pathogen coenrichment with multiple target DNA identification to reliably and effectively detect the three major pathogens in food.
To achieve high detection specificity and avoid competition in the multiple target amplifications, the selection of pathogen-specific target genes and the design of compatible PCR primers and probes are the critical steps. Based on literature searches and sequence homology analyses, the Salmonella invA gene, the E. coli O157 rfbE gene, and the L. monocytogenes hlyA gene were chosen as detection targets. invA is a virulence gene encoding an invasion protein and exclusively exists in almost all Salmonella spp. It has been proven to be Salmonella specific in this study and others (Rahn et al., 1992). rfbE, which is involved in the biosynthesis of the lipopolysaccharide O157 antigen, is conserved in both E. coli O157:H7 and E. coli O157 with non-H7 flagellar antigens (Bilge et al., 1996). As Shiga-toxigenic E. coli O157 is the most frequently isolated pathogenic E. coli strain from humans and the predominant cause of hemolytic uremic syndrome, the identification of E. coli O157 serotype using rfbE as a target was carried out in this study. hlyA, encoding a major virulence factor called listeriolysin O, is the most commonly used target for detecting Listeria monocytogenes (Nogva et al., 2000). DNA sequence analysis showed that the ilo gene encoding ivanolysin O in L. ivanovii and the lso gene encoding seeligerolysin O in L. seeligeri shared 78% and 77% sequence homologies to hlyA, respectively (Haas et al., 1992). To discriminate the pathogenic L. monocytogenes from the nonpathogenic Listeria spp., multiple sequence alignment of the hlyA, ilo, and lso genes was performed and a sequence divergent region in hlyA was selected as a detection target for L. monocytogenes in this study. Using the unique DNA sequences in the selected target genes, three new sets of PCR primers and TaqMan probes were designed. The specificity of all the primers and probes were verified by BLAST against the entire nucleotide database in GenBank and was confirmed in both conventional PCR (data not shown) and multiplex real-time PCR experiments. All of the results from testing the pure bacterial cultures and meat samples demonstrated the high specificity and compatibility of the primer/probe sets for coamplifications of the Salmonella spp., E. coli O157, L. monocytogenes targets, and IAC in the same reaction.
Including IAC in PCR detection of microbial pathogens in food samples is important to ensure data reliability (Hoorfar et al., 2003, 2004). Currently, only a few published real-time PCR detection methods contained the IAC as a false-negative indicator (Long et al., 2008; O'Grady et al., 2008; Sohni et al., 2008). The coamplification of the IAC with target DNA in the same reaction tube requires a unique DNA sequence, a distinctive detection signal, as well as evidence that no inhibition on target amplification occurs. In this study, a novel IAC utilizing a human adenovirus sequence was designed and thoroughly evaluated in the multiplex real-time PCR assay. At an optimized IAC concentration, neither cross-reaction with other DNAs nor inhibitions of the target amplifications were observed, which suggests that the IAC could be reliably used under these conditions.
Using the optimized concentration of 200 nM for each set of primers and probes, and the TaqMan Gene Expression Master Mix with its recommended standard thermal profile, the multiplex real-time PCR assay described herein was able to detect as few as eight, five, and two genome copies of Salmonella, E. coli O157, and L. monocytogenes, respectively, in pure cultures (Table 3). In artificially contaminated ground beef, the multiplex real-time PCR combined with the multipathogen selective enrichment was able to simultaneously enrich and detect the three pathogens in the presence of background microflora, which were determined by plating enriched samples on BHI agar plates (data not shown). Detection limit was achieved at 18 CFU/10 g (initial inoculum level) ground beef or even lower depending on the microorganisms and enrichment time. This was comparable to the sensitivity of conventional multiplex PCR assay of frozen food (Kawasaki et al., 2008) and displayed a 10- to 1000-fold improvement from the PCR analysis of wheat grain samples (Kim et al., 2006). Comparing the quantitative data of viable cell counts and genome copy numbers after enrichment of the spiked beef, we observed some difference in the numbers of CFU/mL versus genomic DNA copy/mL (Table 4). As the cell counting method determines only the number of culturable cells that can grow on the pathogen selective agar plates, whereas the PCR method detects the total number of genome copies from both culturable and nonculturable cells, the genome copy of a sample containing nonculturable cells could be higher than the CFU of the same sample. The nonquantitative results of testing 26 naturally contaminated meat samples by the multiplex real-time PCR and culture-based methods agreed well with each other, suggesting that the multiplex real-time PCR is a reliable method for simultaneously detecting contamination with Salmonella spp., E. coli O157, and L. monocytogenes in food.
In terms of detection speed of multiple pathogens, the entire process of the multiplex assay from sample enrichment to data analysis can be completed in 24 h. The effectiveness of the detection was significantly improved from traditional culturing methods, which require 5–7 days to analyze a single species.
The multiplex real-time PCR assay developed herein offers simplicity, speed, and sensitivity in detecting Salmonella spp., E. coli O157, and L. monocytogenes simultaneously in food. However, the assay limits the number of targets that can be analyzed in each multiplex PCR. As only three gene targets were included in the assay and each of them was used for identifying one of the species, the detection capacity and discriminatory power of the assay are relatively low compared with microarray-based detections. In particular, the rfbE gene used in the assay is specific to the O157 antigen in E. coli. It is insufficient to use rfbE only to discriminate Shiga toxin–producing E. coli (STEC) E. coli O157:H7 from nonpathogenic E. coli O157. To exclusively identify STEC E. coli O157:H7, additional genotype information, including fliC (encoding H7 flagellin), stx1 (encoding Shiga toxin1), stx2 (encoding Shiga toxin2), and eae (encoding intimin), needs to be provided from other assays. To verify PCR results, culture-based pathogen isolation followed by phenotype characterization should also be performed.
In conclusion, a rapid and sensitive multiplex assay was developed for simultaneous detection of Salmonella spp., E. coli O157, and L. monocytogenes in meat products by combining a multipathogen enrichment strategy with a multiplex real-time PCR detection. Considering the sensitivity, specificity, and effectiveness, the multiplex assay developed herein appears to be a promising tool for high-throughput screening of a large number of food samples that require either single or multiple pathogen detection.
Footnotes
Acknowledgments
This research was supported by the Agriculture Research Service, U.S. Department of Agriculture, the National Natural Science Foundation of China (grant no. 30972485), and the Science and Technology Commission of Shanghai Municipality (grant nos. 08391911000, 08142200700, and 08DZ0504200). The authors thank Dr. George Paoli for kindly providing the bacterial strains, and Dr. Peter Irwin and Sue Reed for critically reviewing the manuscript.
Disclosure Statement
No competing financial interests exist.