
Abstract
Salmonella sp., Listeria monocytogenes, and Escherichia coli O157:H7 are foodborne pathogens capable of causing serious gastrointestinal illness. We previously described simultaneous detection of these pathogens by multiplex polymerase chain reaction (PCR) in 44 types of spiked food samples, including meat, produce, fish, and dairy products, targeting genes specific for each pathogen. Based on the previous work, a multiplex real-time PCR assay using fluorescent probes was developed to detect and accurately quantify Salmonella sp., L. monocytogenes, and E. coli O157:H7 in ground pork samples. The detection sensitivity for this method was 2.0 × 102 CFU/mL for each pathogen, and the quantification range was 102–107 CFU/mL with a high correlation coefficient (R 2 > 0.99) and high PCR efficiency (84.2% to 99.2%). When this protocol was used for the detection of each of the pathogens in spiked pork samples, one cell per 25 g of inoculated sample after enrichment for 20 h could be detected within 24 h. As a result, this multiplex real-time PCR assay will be valuable as a screening method for foods contaminated with these pathogens.
Introduction
S
Conventional culture-based methods are labor intensive, time consuming, and costly; therefore, more rapid methods for specific detection and identification of these pathogens is required to eliminate the potential for contaminated food reaching the consumer. In recent years, rapid and automated assays have been developed and validated employing a wide range of commonly used microbiological methods. Polymerase chain reaction (PCR)-based methods have been developed for rapidly detecting pathogenic bacteria in foods and other types of samples with high sensitivity and specificity. Thus, PCR-based assays are currently more widely used for screening a variety of pathogens in different foods and other types of samples (Croci et al., 2004; Fratamico and Kawasaki, 2008).
Multiplex PCR systems, carried out in a single tube for simultaneous detection of more than one bacterial species, have been investigated because they offer the advantages of savings in time and cost (Wagner et al., 2000; Kawasaki et al., 2005; Park et al., 2006). We developed a multiplex PCR method capable of determining the presence of Salmonella sp., L. monocytogenes, and E. coli O157:H7 directly from enrichment cultures (Kawasaki et al., 2005) of food samples. When this method was applied to spiked ground pork samples, the detection sensitivity for each pathogen was 1 CFU/25 g of inoculated sample after enrichment for 20 h (Kawasaki et al., 2005). Excellent agreement between results of the multiplex PCR assay and the conventional culture method was obtained using naturally contaminated meat samples. Moreover, we evaluated the sensitivity of the multiplex PCR method with various spiked frozen and nonfrozen food samples and with various naturally contaminated foods. The assay was capable of detecting as low as 5 CFU/25 g of each pathogen in more than 40 types of food samples after enrichment in No. 17 broth (Kawasaki et al., 2009).
Relying on the use of gel electrophoresis for detection of products in multiplex PCR systems is a problem if automation of the assay or rapid screening of samples is required. Further, the number of samples that can be analyzed at one time is also limited, and the ethidium bromide used to stain the agarose gels is a hazard for routine use in food laboratories. On the other hand, real-time PCR assays employing various types of fluorescence systems allow direct detection of product, without gel electrophoresis and staining steps (Oberst et al., 1998; Kimura et al., 1999), and the detection limit is generally in the range of 102–103 CFU/mL. In this study, a real-time multiplex PCR detection method for Salmonella sp., L. monocytogenes, and E. coli O157:H7 was developed, which was used for specific detection and quantification of each pathogen in ground pork samples.
Materials and Methods
Bacterial strains and culture conditions
The bacterial strains used were Salmonella Enteritidis IFO3313, L. monocytogenes ATCC 49594, and E. coli O157:H7 ATCC 43894. Bacterial strains were grown overnight at 37°C with rotary shaking in trypticase soy broth (BBL, Cockeysville, MD) containing 0.6% yeast extract, unless otherwise stated. For determining the number of viable cells, serial decimal dilutions of cultures in phosphate-buffered saline were plated onto trypticase soy agar (BBL) for each bacterium and incubated at 37°C for 48 h before enumeration. Seventeen target strains (2 strains of Salmonella Enteritidis, 2 strains of Salmonella Typhimurium, 9 strains of L. monocytogenes, and 4 strains of E. coli O157:H7) and 15 nontarget strains (Listeria ivanovii, Listeria welshimeri, Listeria innocua, Listeria seeligeri, Pseudomonas fragi, Citrobacter freundii, 2 Leuconostoc mesenteroides strains, 2 Lactobacillus viridescens strains, and 5 non-O157:H7 E. coli strains) were grown at 37°C overnight in trypticase soy broth medium and were used for specificity testing.
Specificity and sensitivity of the multiplex PCR assay using DNA from pure cultures
Specificity and sensitivity testing using the above strains was performed by multiplex real-time PCR using chromosomal DNA purified from the bacteria by using standard methods (Sambrook et al., 1989). The concentration of chromosomal DNA diluted in TE buffer was determined using a spectrophotometer (Ultrospec 3300 pro; GE Healthcare UK, Amersham Place, Little Chalfont, England). Two nanograms, 200 pg, 20 pg, 2 pg, 200 fg, and 20 fg of DNA was used as the PCR template to determine specificity and sensitivity of the assay.
Enrichment, DNA extraction, and multiplex real-time PCR conditions
The No. 17 enrichment medium (10 g tryptose, 5 g of beef extract, 5 g of yeast extract, 15 g of monopotassium phosphate, 7 g of disodium phosphate, 5 g of sodium chloride, and 0.5 g of dextrose), which was described previously (Kawasaki et al., 2005), was used for simultaneous growth of Salmonella Enteritidis, L. monocytogenes, and E. coli O157:H7 in this study. This medium allows for adequate growth of the three pathogens for detection by PCR. DNA was extracted using the lysis-guanidine isothiocyanate (GuSCN) method also, as described previously (Kawasaki et al., 2005). Briefly, aliquots (1 mL) from the enrichments were transferred to microcentrifuge tubes, and the cells were collected by centrifugation. The pellets were resuspended in 200 μL of enzyme solution containing 1 mg/mL each of acromopeptidase and lysozyme in TE buffer. After incubation for 1 h at 37°C, the solution was mixed with 300 μL of 4 M GuSCN solution containing 2% (wt/vol) Tween 20. A portion (400 μL) of the supernatant was transferred to a new tube containing 400 μL of 100% isopropanol. After mixing, the mixture was centrifuged for 10 min at 15,000 g, and the resulting DNA pellet was rinsed with 75% isopropanol. The pellet was then dissolved in 160 μL of distilled water with heating for 3 min at 70°C. Prior to use, the template DNA solution was centrifuged for 5 min at 15,000 g to further remove water-insoluble impurities. Two microliters of the solution was used as template for the PCR.
The multiplex PCR mixture described previously (Kawasaki et al., 2005) was modified by the addition of probes labeled with different fluorophores (Table 1). The primers targeted a Salmonella species-specific sequence within a 1.8-kb HindIII DNA fragment (Tsen et al., 1994), the L. monocytogenes hlyA gene (Furrer et al., 1991), and the E. coli O157:H7 eaeA gene (Sharma et al., 1999). The probes were labeled at the 5′ end with the fluorescent reporter dye and at the 3′ end with a quencher dye. For Salmonella, the detection probe was labeled with FAM and BHQ1, the L. monocytogenes detection probe was labeled with TET and BHQ1, and the E. coli O157:H7 detection probe was labeled with BODIPY-TMR and BHQ2. PCR was performed in a total volume of 50 μL consisting of 2 μL of template DNA and 48 μL of PCR master mix composed of 1 × PCR buffer II, 5.0 mM MgCl2, 120 nM concentrations of each Salmonella detection primer, 100 nM concentrations of each L. monocytogenes detection primer, 80 nM concentrations of each E. coli O157:H7 detection primer, 200 μM dATP, dCTP, and dGTP, 400 μM dUTP, 0.025 U of AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA), 0.05 U of uracil-N-glycosidase (AmpErase UNG; Applied Biosystems), and 25 nM concentrations of each fluorescence detection probe with an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). The thermal cycling setup had a carryover treatment using uracil-N-glycosidase at 50°C for 2 min and initial denaturation at 95°C for 10 min, which was followed by 40 cycles of denaturation (95°C, 20 sec), annealing (60°C, 30 sec), and extension (72°C, 30 sec) and a final step of 7 min at 72°C. The expected sizes of the PCR products for Salmonella sp., L. monocytogenes, and E. coli O157:H7 were 375, 234, and 120 bp, respectively.
PCR, polymerase chain reaction.
For real-time PCR, the ABI PRISM 7900 Sequence Detection System uses a software algorithm to calculate dRn, which is the change in the fluorescence. The fluorescence signal was normalized by dividing the emission intensity of the reporter dyes (FAM, TET, and BODIPY-TMR) by the emission intensity of a reference dye (ROX) to obtain a ratio defined as dRn (dRnFAM, dRnTET, and dRnBoTMR) for a given reaction tube. The threshold dRn was set at 10 times the standard deviation above the mean baseline fluorescence calculated for PCR cycles 3 to 15. A reaction was considered positive if the dRn curve exceeded the threshold at the completion of 40 cycles. For quantification of the PCR products, a fluorescence threshold was manually set (dRn = 0.2) across all samples in the experiment such that it bisected the exponential phase of the fluorescent signal increase. The cycle threshold (Ct) was defined as the cycle number at which a sample's dRn fluorescence crossed the threshold. Data collection and multicomponent analysis were performed with the Sequence Detection Software 2.0 (Applied Biosystems) program. In addition, the PCR products were verified by agarose gel electrophoresis and staining with ethidium bromide.
Standard curve construction
Serial dilutions were made from overnight cultures of Salmonella Enteritidis, L. monocytogenes, and E. coli O157:H7, and DNA was extracted as described above from 2 × 106, 2 × 105, 2 × 104, 2 × 103, and 2 × 102 cells/mL, which was then used as the PCR template for determining the standard curve. Each dilution was tested in duplicate. The ABI PRISM 7900 Sequence Detection System software automatically calculates the standard curve for each run based on the Ct for each standard. Based on these values, a linear regression line was plotted, and the resulting equation was used to calculate the log10 CFU/mL for determination of sensitivity, standard curve, and amplification efficiency. The formula from which the amplification efficiency was calculated is e = 10−1/s − 1, where s is the slope of the standard curve.
Multiplex PCR evaluation in spiked ground pork samples
Ground pork was purchased from a local supermarket and was immediately transported in an insulated cooler at 4°C to the laboratory for inoculation and analysis on the same day. One milliliter of a mixed culture containing Salmonella Enteritidis, L. monocytogenes, and E. coli O157:H7 (100, 101, and 102 CFU of each pathogen) was mixed into 25 g of ground pork, and 225 mL of No. 17 medium was added. The samples were then pummeled using a Stomacher for 2 min and then incubated at 35°C. After 20 h, 1 mL of the enrichment was used for DNA recovery by the lysis-GuSCN method as described earlier and then subjected to the multiplex real-time PCR assay. The ABI PRISM 7900 software was used to determine if the samples were positive or negative for the target pathogens based on the fluorescence intensity.
Results
Probe design and detection limit for the multiplex PCR assay
The multiplex PCR method was capable of simultaneously determining the presence of Salmonella sp., L. monocytogenes, and E. coli O157:H7 by targeting a Salmonella-specific DNA region, the L. monocytogenes hlyA gene, and the E. coli O157:H7 eaeA gene (Kawasaki et al., 2005) using the primers and probes shown in Table 1. Each probe, targeting regions between the two primer binding sites, was labeled with a different fluorescent dye (FAM [520 nm], TET [540 nm], and BODIPY-TMR [570 nm] for Salmonella sp., L. monocytogenes, and E. coli O157:H7, respectively) at the 5′ end and with a quencher dye (BHQ1, BHQ1, and BHQ2, respectively) at the 3′ end. Table 2 shows that using a mixture of DNA from the three pathogens as template in the multiplex PCR assay, the detection limit was 200 fg for each pathogen per PCR. The presence of the products with expected sizes was confirmed by agarose gel electrophoresis, and no nonspecific products were obtained (data not shown).
The specificity of real-time multiplex PCR
The specificity of the real-time multiplex PCR assay was evaluated using 17 target strains (4 strains of Salmonella sp., 9 strains of L. monocytogenes, and 4 strains of E. coli O157:H7) and 14 nontarget strains. The fluorescence signal with FAM was only detected for Salmonella sp., and TET and BODIPY-TMR only with L. monocytogenes and E. coli O157:H7, respectively. None of the nontarget strains produced a fluorescence signal in the multiplex PCR assay.
Assay sensitivity and standard curve
To construct the standard curve for each pathogen, DNA from 10-fold serial dilutions of target pathogen cells (ranging from 107 to 102 cells/mL) was evaluated. Testing of DNA from samples containing different concentrations of Salmonella Enteritidis, L. monocytogenes, and E. coli O157:H7 showed that PCR gave a positive result using samples containing approximately 2 × 102 cells/mL of each of the target pathogens in less than 40 cycles. The standard curves showed a liner relationship between log input of cells and the Ct number (Fig. 1a–c). The slope of these curves were in the range of −3.341 to −3.769, and the correlation coefficient (r 2) after the linear regression was above 0.99 (Table 3).
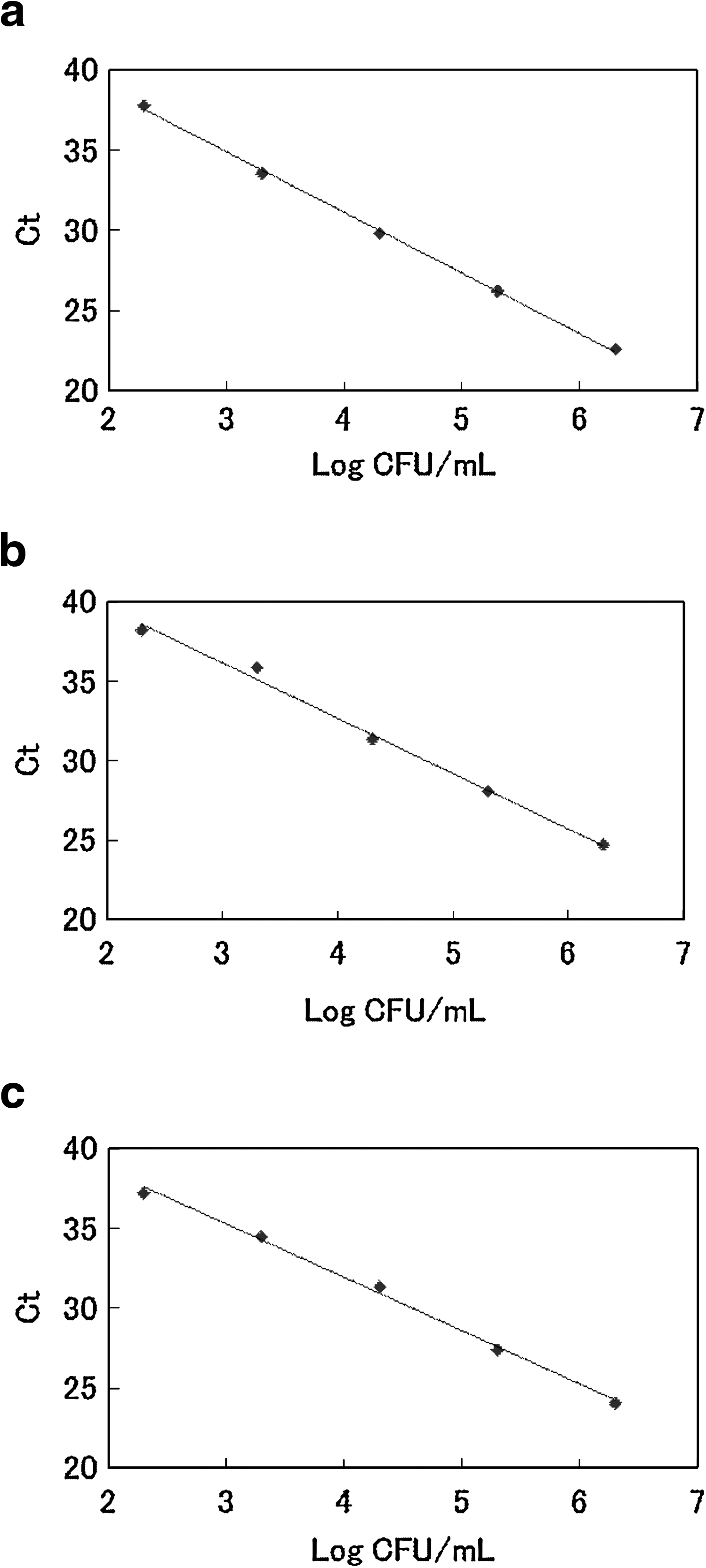
Standard curves showing the relationship between Ct and log10 CFU/mL for the multiplex real-time polymerase chain reaction assay. Ct, cycle threshold.
The standard curve was constructed from the Ct versus log10 CFU of each pathogen.
The amplification efficiency was calculated from the following formula: e = 10−1/s − 1, where s is the slope of the standard curve.
Ct, cycle threshold.
Evaluation of the multiplex real-time PCR assay with spiked ground pork samples
The sensitivity of the multiplex PCR assay was evaluated using ground pork spiked with the three pathogens simultaneously at 1, 10, and 100 CFU/25 g of each pathogen. In the contaminated ground pork samples, Salmonella Enteritidis, L. monocytogenes, and E. coli O157:H7 were all detected by this multiplex real-time PCR assay at concentrations as low as 1 CFU/25 g after enrichment in No. 17 broth for 20 h (Table 4). Detection was also possible after 16 h of enrichment (data not shown).
Discussion
In our previous study, we developed a multiplex PCR method for simultaneous detection of Salmonella sp., L. monocytogenes, and E. coli O157:H7 and evaluated the method with 44 types of food. The detection limit was as few as 1 CFU/25 g of each of the target pathogens (Kawasaki et al., 2005, 2009). Moreover, the detection rate was higher than that of the conventional culture method for all frozen samples (at −20°C for 2 weeks or 2 months) and for naturally contaminated samples examined. However, this multiplex PCR method involves the use of gel electrophoresis to determine the presence of amplicons, which requires skill and is laborious, time consuming, and hazardous. In this study, we designed a multiplex real-time PCR assay for detection and quantification of the target pathogens using different fluorescence probes targeting each pathogen. The real-time multiplex PCR assay is more rapid and makes it possible to analyze many samples simultaneously.
For simultaneous growth of Salmonella sp., L. monocytogenes, and E. coli O157:H7, the No. 17 broth medium was used in this study. No. 17 broth does not contain selective agents, and favorable pH conditions are maintained by limiting the carbohydrate content, which is particularly helpful for L. monocytogenes cultivation. Omiccioli et al. (2009) also described simultaneous detection of pathogens from milk samples with No. 17 broth that was modified by removing dextrose. However, it was found that removing dextrose from No. 17 broth delays the recovery of injured cells (data not shown); therefore, in this study, No. 17 broth contained dextrose.
Initially, we evaluated a multiplex real-time PCR assay using the intercalating dye, SYBR Green, for direct real-time PCR detection with post-PCR analysis of dissociation curves for each PCR amplicon. However, it was difficult to analyze the SYBR Green fluorescence data because the generated primer dimers resulted in high background fluorescence intensities (data not shown). Therefore, in this study, specific fluorescence probes for each pathogen were used in the real-time multiplex PCR assay. To select suitable reporter dyes for multiplex real-time PCR detection, the spectral data were measured with 12 types of fluorophores (FAM, Colflour540, TET, JOE, VIC, HEX, Colflour560, Alexa546, BODIPY-TMR, NED, TAMRA, and ROX). Finally, FAM, TET, and BODIPY-TMR were selected as the reporter dyes, because the wavelengths of maximum excitation intensity did not result in interference in the fluorescence readings by the ABI PRISM 7900 instrument.
Using 10-fold dilutions of target DNA from each pathogen, the sensitivity of the multiplex real-time PCR assay was 200 fg/PCR (Table 2). The advantage of the multiplex real-time PCR assay is that there is a wide dynamic range, making quantification of the target pathogens possible. The relationship between Ct values and target DNA concentrations gave a good correlation (r 2 > 0.99). The amplification efficiency was 84.2% (Salmonella sp.; 375-bp product), 93.8% (L. monocytogenes; 234-bp product), and 99.2% (E. coli O157:H7; 120-bp product) for each pathogen in the multiplex PCR assay (Table 3), and the standard curve had a dynamic range from 102 to 107 cells/mL. Generally, there is a reverse correlation between the amplification efficiency and the size of PCR product; therefore, it may be necessary to adjust for each amplification efficiency of PCR product for construction of multiplex PCR condition.
Moreover, one cell per 25 g of inoculated ground pork sample could be detected by the multiplex real-time PCR assay after enrichment in No. 17 broth for 20 h (Table 4), which was similar to the sensitivity of the PCR assay developed previously (Kawasaki et al., 2009). The application of real-time PCR for pathogen detection of Yersinia enterocolitica in ground beef, chicken carcasses, milk, cold smoked sausage, carrots, and fish was described by Lambertz et al. (2008), who showed that it was a useful method for screening by using a commercially available DNA extraction kit for DNA purification from the samples. In this study, artificially contaminated ground pork was used for the multiplex real-time PCR, and the assay was sensitive and specific for determining the absence/presence of Salmonella sp., L. monocytogenes, and E. coli O157:H7 (Table 4). In our previous report, a simple DNA extraction method (lysis-GuSCN method) was used for detecting the pathogens from 44 different foods. DNA extraction methods using columns, magnetic beads, or other techniques have also been used for PCR detection from foods. Many of these protocols may be more suitable for liquid foods, such as milk, and may be difficult to apply to more complex foods, such as meat. In addition, the cost of column-based (e.g., DNeasy Blood and Tissue kit) or magnetic bead-based (e.g., Dynabeads, PATHATRIX) systems, which are commercially available, is relatively high, and thus, these techniques may not be suitable for routine testing by the food industry. The lysis-GuSCN method does not require any column for the extraction of DNA from food samples. A comparison of the lysis-GuSCN method to column-based (Elizaquivel and Aznar, 2008) or magnetic bead-based extraction systems (Uttendaele et al., 2000; Warren et al., 2007) showed that it was simpler, and more reproducible PCR results were obtained (data not shown).
In conclusion, the real-time multiplex PCR assay, which included enrichment (20 h) of ground pork samples, DNA extraction (<2 h), and real-time PCR amplification and detection (<2 h), exhibited high selectivity and sensitivity and is thus a robust tool for testing many samples for Salmonella sp., L. monocytogenes, and E. coli O157:H7 simultaneously within 24 h. This method is suitable for quantitative detection down to levels of approximately 102 cells/mL as well. This multiplex real-time PCR assay could be valuable as a screening method for foods contaminated with Salmonella sp., L. monocytogenes, and E. coli O157:H7 and could potentially also be useful for identifying the sources of foodborne outbreaks.
Footnotes
Acknowledgments
This work was supported in part by a grant (Research project for ensuring food safety from farm to table, DI-7107) from the Ministry of Agriculture, Forestry, and Fisheries of Japan. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Disclosure Statement
No competing financial interests exist.