
Abstract
A two-step real-time SYBR Green I multiplex polymerase chain reaction (PCR) assay with melting curve analysis was developed for rapid detection of 19 Salmonella serotypes frequently encountered in humans, animals, and animal-associated meat products within the European Union. The first-step single-tube reaction (Multiplex PCR I), consisting of five primer pairs, classified an initial test panel of eight Salmonella serotypes into five groups on the basis of characteristic amplicon melting temperatures produced by each strain. Following designation into groups, two subsequent triplex reactions (Multiplex PCR II-G1 and II-G3) allowed for further identification of five Salmonella serotypes by their melting peak temperatures. Primers for serotype differentiation were designed to target the genes encoding either phase 1 and 2 flagellar antigens fliC and fljB or unique serotype-specific loci. In addition, the assay simultaneously screened for the presence of the ampicilin-amoxicillin, chloramphenicol-florfenicol, streptomycin-spectinomycin, sulfanomides, and tetracycline (ACSSuT)-type multidrug resistance pattern, indicated by the floR gene, and for the Salmonella virulence plasmid encoded by the svp operon in Salmonella serotype Typhimurium. The established multiplex assays were successfully tested on 97 isolates, comprising 37 distinct Salmonella serotypes and 12 non-Salmonella strains. The two-step assay correctly detected 19 of 37 Salmonella serotypes and all non-Salmonella strains produced negative results. Of the 19 serotypes detected in the assays, 7 serotypes, including Salmonella serotypes Ohio, Goldcoast, Livingstone, Kedougou, Enteritidis, Kentucky, ACSSuT-type Salmonella serotype Typhimurium DT104 and DT104b, as well as non-ACSSuT-type Salmonella serotype Typhimurium strains, were definitively identified. The developed multiplex real-time SYBR Green I PCR assay represents a more rapid and reliable method for identification of large numbers of Salmonella serotypes prevalent throughout the European Union than assays using phenotypic serotyping methods.
Introduction
Serotyping according to the Kauffmann-White serological scheme is currently the most common method to identify and discriminate between Salmonella enterica subsp. enterica isolates. Serotype identification is based on the O (surface polysaccharide) and H (flagellar) antigens present in the cell wall of the organism (Kim et al., 2006). Genes encoding the O antigens are located in the rfb cluster, whereas phase 1 (H1) and phase 2 (H2) flagellar antigens are alternatively expressed by a phase variation mechanism, involving fliC and fljB, respectively (Echeita et al., 2002). Serotype determination by the slide agglutination method has major drawbacks as it is labor intensive, requires the use of >150 anti-sera, and usually takes at least 3 days to obtain results (Echeita et al., 2002). In addition, interpretation of results may be subjective. Traditional serotyping often requires a strain phase reversal as usually only one flagellar antigen is expressed at a time delaying full identification of both flagellar antigens (Echeita et al., 2002). Monophasic strains express only one flagellar antigen and account for a large proportion of misidentified strains (Hendriksen et al., 2009). Salmonella 4,[5],12:i:-, which is most likely a monophasic variant of Salmonella Typhimurium (Echeita et al., 2001), accounted for the greatest number of incorrectly typed isolates in 2006, according to the Global Salm-Surv external quality assurance system launched by the World Health Organization. Difficulties in the detection of the phase 2 flagellar antigen and discrimination between particular H antigen complexes also resulted in a significant number of errors in serotyping (Hendriksen et al., 2009).
Recently, flagellar genes have been suggested as useful targets to distinguish between various Salmonella serotypes at a molecular level. Molecular approaches to Salmonella serotyping have been employed in several studies and included methods such as TaqMan probe-based real-time multiplex polymerase chain reaction (PCR) assays (O'Regan et al., 2008; Woods et al., 2008) and conventional multiplex PCR (Echeita et al., 2002; Kim et al., 2006; Herrera-León et al., 2007; Hong et al., 2008; Lim and Thong, 2009). The use of real-time PCR methods offers considerable advantages over conventional PCR, as no post-PCR processing is necessary to observe amplification products generated throughout the reaction. Handling of PCR products is time consuming and may introduce cross-contamination compared to real-time PCR, which uses closed-tube detection and allows for faster cycling times as PCR product amplification and analysis occur simultaneously (Ririe et al., 1997; Yoshitomi et al., 2006). In addition, a real-time PCR-based approach has been proposed as an alternative to traditional serotyping as it is a precise, reproducible, and rapid method with capacity for high throughput (Heid et al., 1996; Kim, 2001; Olsen et al., 2009). The use of SYBR Green I dye is of particular advantage as it directly detects double-stranded DNA during amplification enabling post-PCR melting curve analysis for rapid identification of desired target genes (Ririe et al., 1997).
Our objectives were to develop and validate a multiplex real-time SYBR Green I-based PCR assay with melting curve analysis to rapidly identify Salmonella serotypes frequently isolated from humans and animals in the EU. This assay is particularly suitable for screening large numbers of Salmonella isolates.
Materials and Methods
To cover a broad spectrum of Salmonella serotypes and to allow for differentiation, identification of serotypes was based on three real-time PCR assays consisting of an initial reaction step termed Multiplex PCR I, allowing designation of isolates into five groups, and two subsequent reactions named Multiplex PCR II-G1 and G-3 (Fig. 1).

Overview of the Salmonella serotype identification scheme indicating the target genes and the initial test panel of Salmonella serotypes used for development of the real-time Multiplex PCR I, II-G1, and II-G3 assays. aFurther conventional serotyping required for complete identification. bIdentification of ACSSuT-type Salmonella Typhimurium strains (resistant to ampicilin-amoxicillin, chloramphenicol-florfenicol, streptomycin-spectinomycin, sulfanomides, and tetracycline). No further differentiation step required. cIdentification of Salmonella Enteritidis, no further differentiation step required. dStrain no. L6, isolate received from Backweston Laboratory Complex, Ireland. eStrain no. S10-001478, isolate received from Backweston Laboratory Complex, Ireland. fStrain no. D8, isolate received from Laboratory of Food Microbiology and Infectious Diseases, UCD, Ireland. gStrain no. S10-000412, isolate received from Backweston Laboratory Complex, Ireland. hStrain no. A22, isolate received from Backweston Laboratory Complex, Ireland. iStrain no. H21, isolate received from Backweston Laboratory Complex, Ireland. jStrain no. R42, isolate received from Laboratory of Food Microbiology and Infectious Diseases, UCD, Ireland. kStrain no. F6, isolate received from Backweston Laboratory Complex, Ireland. lStrain no. S09-002259, isolate received from Backweston Laboratory Complex, Ireland. PCR, polymerase chain reaction; ACSSuT, ampicilin-amoxicillin, chloramphenicol-florfenicol, streptomycin-spectinomycin, sulfanomides, and tetracycline.
Bacterial strains and growth conditions
An initial test panel of eight Salmonella serotypes were used for the development of the three real-time multiplex PCR assays described in this study. These strains were chosen because they are the most frequently reported Salmonella serotypes in the EU (Anonymous, 2010) and included Salmonella Bredeney, Salmonella Ohio, Salmonella Goldcoast, Salmonella Livingstone, Salmonella Derby, Salmonella Typhimurium (non-ACSSuT-type Salmonella Typhimurium and ACSSuT-type Salmonella Typhimurium DT104b), Salmonella Kentucky, and Salmonella Enteritidis (Fig. 1). Upon establishment of the PCR assays, an expanded panel of 85 Salmonella isolates representing 37 serotypes frequently encountered in the EU was used to determine accuracy and specificity of these assays (Table 1). In addition, 12 non-Salmonella strains that show either phylogenetic relationships to the fliC gene (Macnab, 1992) or that produce similar colony appearance on Xylose Lysine Decarboxylase agar were tested to validate the specificity of the established multiplex real-time PCR assays (Table 2). Salmonella strains were cultured on Xylose Lysine Decarboxylase (Lab M Ltd., Lancashire, United Kingdom); non-Salmonella strains were grown on blood agar (Oxoid LTD., Hampshire, England) at 37°C for 24 h.
H1, phase 1 flagellar H-antigen.
H2, phase 2 flagellar H-antigen.
Isolate received from UCD Centre for Food Safety, Dublin, Ireland.
Isolate received from Laboratory of Food Microbiology and Infectious Diseases, UCD, Ireland.
DNA template preparation
Crude DNA was isolated from pure cultures of bacteria. A single colony was suspended in 50 μL of nuclease-free water in a 1.5 mL microcentrifuge tube. The suspension was heated at 100°C for 5 min and centrifuged at 12,000 g for 2 min. One microliter of the supernatant was subsequently used for the PCR.
Primer design and optimization
Genes chosen for serotype identification were based on antigenic properties of the phase 1 and phase 2 flagellar genes fliC and fljB as well as unique serotype-specific loci, including sdr, floR, and spv (Fig. 1). All primer pairs designed for this study are listed in Table 3. Primer design was based on DNA sequences retrieved from the GenBank database. The fliC and fljB primers were designed to target the central, hypervariable regions that encode the surface-exposed variable proportion of these bacterial flagellins (McQuiston et al., 2004). Regions displaying the greatest degree of polymorphism were chosen as determined by multiple alignments using ClustalW2 (
Primer name as it appears in the text.
Calculated PCR amplicon GC content (%) with Oligo Calc (
GenBank accession number used for design of forward and reverse primer.
PCR, polymerase chain reaction.
Primers were optimized to a final concentration of 300 nM for the combinations FliC/FljB-l-F and FliC/FljB-l-R; FliC-g-F and FliC-g-R; FliC-i-R, FloR-R, Sdr-F, and Sdr-R; FliC-d-F and FliC-d-R; FliC-r,i-F and FliC-r,i-R; FliC-b-F and fliC-b-R; Spv-F and Spv-R; FljB1-z6-F and FljB1-z6–R; and FljB-1,2/1,5-F and FljB-1,2/1,5-R; and 150 Nm for FliC-i-F and 50 nM for FloR-F. Each primer pair was tested individually in a singleplex reaction on all 37 Salmonella serotypes used in this study to verify its specificity before it was combined in a multiplex reaction. All primers were specifically designed to produce unique amplicon sizes, allowing for clear discrimination after melting curve analysis.
SYBR Green I real-time PCR
The SYBR Green I amplification mixture with a final volume of 25 μL for all real-time PCR assays included 1× Power SYBR® Green I PCR Master Mix (Applied Biosystems, Warrington, United Kingdom), sterile Rnase-free water, forward and reverse primers in the appropriate concentration, and 1 μL crude DNA extract. PCRs were performed on an Applied Biosystems 7300 Real-Time PCR system (Applied Biosystems) using the following cycling conditions: 1 cycle at 50°C for 2 min, followed by 1 cycle at 95°C for 10 min, 40 cycles of denaturation for 15 s at 95°C, and an annealing/extension step at 60°C for 1 min. Melting curve data were generated using 1 cycle at 95°C for 15 s, 60°C for 60 s, 95°C for 15s, and 60°C for 15 s, while recording fluorescence as a function of temperature. A negative control without target DNA (No template control [NTC]) was included with each run. Each multiplex assay designed for the initial test panel was repeated three times on different days using fresh crude DNA, with each amplification run repeated three times. Positive gene products of the initial test panel were confirmed by conventional gel electrophoresis [3% (w/v)] run in 1× TAE Buffer, and stained with GelRed (Biotium Inc., Hayward, CA). Fragment sizes were determined by comparison with a 25 bp DNA ladder. Screening of the expanded panel (97 strains) used for testing of the established multiplex PCR assays was performed in triplicate. The initial test panel was included as a positive control for each run.
Serotype identification and data analysis
The initial real-time Multiplex PCR I assay comprised five gene targets in one reaction step. The reaction consisted of three serotype-specific primers targeting the central variable regions of the flagellar genes fliC(l,v), fljB(l,w); fliC(f,g), (f,g,s), (f,g,t), (g,s,t), or fliC(i). Two additional primer sets, FloR and Sdr, were also included in the Multiplex PCR I reaction. The floR gene, specific for florfenicol-chloramphenicol resistance, is indicative of multidrug resistance encoded by the SGI-1 island carried by ACSSuT-type strains Salmonella Typhimurium DT104 and DT104b. The Sdr primer set targeting the chromosomally encoded Salmonella difference region of currently unknown function was chosen as a specific marker for Salmonella Enteritidis as it contains loci that are restricted to this serotype (Agron et al., 2001). The second-step assays Multiplex PCR II-G1 and II-G3 consisted each of three gene targets identifying either fliC(d),(r,i),(b) or fljB(z6),(1,2),(1,5) genes of the respective serotypes. In addition, Multiplex PCR II-G3 identified the spv operon, which is encoded on a 94.7 kb serotype-specific virulence plasmid presumably harbored by >90% of clinical Salmonella Typhimurium isolates (Chiu et al., 1999, 2006).
Identification of serotypes possessing one or more gene targets was based on both melting curve analysis of resulting DNA amplicons produced throughout the real-time PCRs and a positive threshold cycle (CT) value (<20 cycles). The identity of the PCR product was confirmed by performing melting curve analysis.
Statistical analysis
One-way analysis of variance followed by Scheffe's post hoc test using Data Desk 6.2 (Data Description, Inc., Ithaca, NY) was employed on data sets to compare differences in melting temperatures and to show statistical significance between the melting peaks produced by either the Multiplex PCR I, Multiplex PCR II-G1, or Multiplex PCR II-G3 assay.
Results
Screening of the initial test panel
Multiplex PCR I
The initial Multiplex PCR I assay comprised five novel primer pairs in one reaction step (Table 3). The FliC/FljB-l primers targeted both the first-phase l,v-antigen of Salmonella Bredeney (encoded by fliC) and the second-phase l,w-antigen (encoded by fljB) present in serotypes Ohio, Goldcoast, and Livingstone. FliC-g simultaneously identified the phase 1 flagellar antigen complex f,g; f,g,s; f,g,t; and g,s,t present in Salmonella Derby. FliC-i targeted the fliC-encoded i-antigen in Salmonella Typhimurium. The gene target floR identified ACSSuT-type strains Salmonella Typhimurium DT104 and DT104b. The Sdr primer specifically targeted Salmonella Enteritidis.
Melting curve analysis of DNA amplicons produced by the Multiplex PCR I assay allowed classification of the initial test panel of eight Salmonella serotypes into five different groups (Groups 1–5) on the basis of their characteristic melting temperatures (Table 4). Average melting temperatures of the group-specific melting peaks were significantly different (p < 0.0001). The serotypes within each group, the primers used, and Tm ranges produced are given in Table 4 and Figure 2A. Unique melting peaks were produced for two Salmonella serotypes/phage type, comprising Salmonella Typhimurium DT104b (Group 4) and Salmonella Enteritidis (Group 5) (Table 4, Fig. 2B). These serotypes were readily identified by the Multiplex PCR I assay and required no further differentiation step, whereas serotypes clustering into Groups 1 and 3 were further differentiated by the subsequent second-step reactions Multiplex PCR II-G1 and Multiplex PCR II-G3.
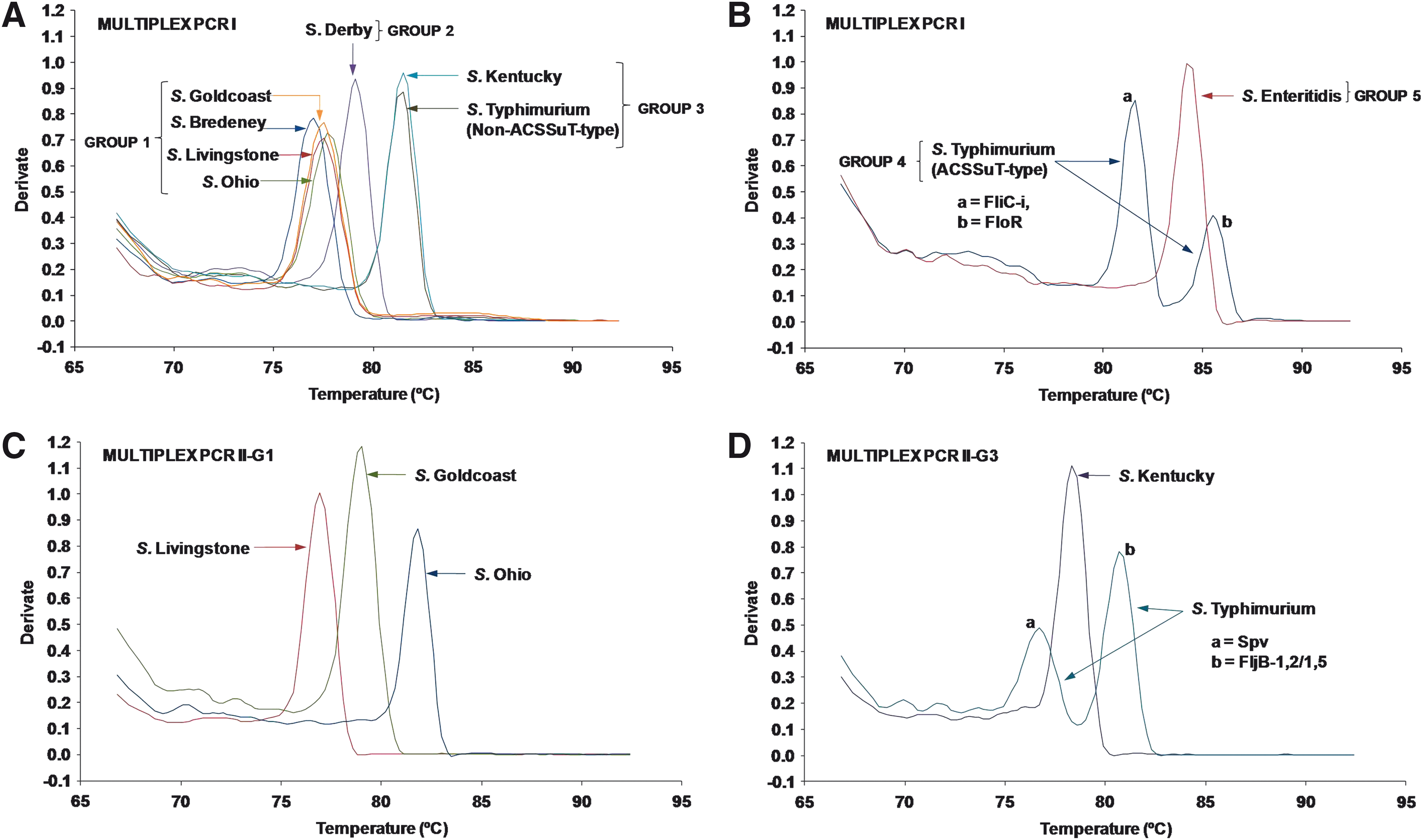
Melting curve analysis of multiplex PCR I
Primer name as it appears in the text.
Average amplicon melting temperature value generated by primer differed significantly from others in Multiplex PCR (p < 0.0001).
Group range is indicated.
Non-ACSSuT-type strain.
Primer combination generated two melting peaks.
ACSSuT-type strain.
ACSSuT, ampicilin-amoxicillin, chloramphenicol-florfenicol, streptomycin-spectinomycin, sulfanomides, and tetracycline.
Multiplex PCR II-G1 and II-G3
Following designation into Group 1 of the five groups differentiated by Multiplex PCR I, Salmonella Ohio, Salmonella Goldcoast, and Salmonella Livingstone were successfully identified by the second-step real-time PCR assay Multiplex II-G1. Serotypes Typhimurium and Kentucky (Group 3) were identified by the Multiplex PCR II-G3 assay.
The Multiplex PCR II-G1 assay consisted of a triplex reaction comprising primer pairs FliC-d, FliC-r,i, and FliC-b targeting the fliC genes of serotypes Livingstone, Goldcoast, and Ohio, respectively (Table 3). Melting curve analysis is depicted in Figure 2C and Tm and CT values are given in Table 4. Finally, the Multiplex PCR II-G3 assay, which consisted of a triplex reaction involving primer pairs FljB(z6), FljB(1,2),(1,5), and Spv discriminated between Salmonella Kentucky and non-ACSSuT-type Salmonella Typhimurium (Tables 3 and 4, Fig. 2D). In addition, this assay targeted the spv operon, thus identifying the presence of the virulence plasmid in Salmonella Typhimurium through generation of a second melting peak.
The melting peaks produced by both Multiplex PCR II-G1 and II-G3 showed statistically significant differences in their average Tm values (p < 0.0001 and p < 0.0001, respectively).
Gel electrophoresis of the Multiplex PCR I, II-G1, and II-G3 reactions
PCR products generated by the Multiplex PCR I, Multiplex PCR II-G1, and Multiplex PCR II-G3 reactions for the initial test panel of eight Salmonella serotypes were observed by agarose gel electrophoresis (Fig. 3). The amplified products displayed the expected size for each of the serotypes, thus confirming melting curve data. Salmonella Typhimurium DT104b generated two PCR amplicons in the Multiplex PCR I assay, which were clearly distinguishable by melting curve analysis resulting from differences in the GC/AT ratio (Fig. 2B). However, these amplicons could not be differentiated on the gel due to close similarity in sizes (142 and 143 bp, respectively) (Fig. 3). Nonspecific, faint bands were observed for the NTCs of each Multiplex PCR assay as well as in the lanes with positive gene targets generated from serotypes in Multiplex PCR I (Fig. 3). Intensity of these bands was most pronounced in the NTCs. The band size of ∼40 bp indicated that these were primer dimers, which most likely resulted from combinations of the large numbers of primers in this reaction. No primer–dimer formation was observed in the NTC of the singleplex reaction performed during primer optimization (data not shown). However, presence of these nonspecific products in samples containing the DNA template did not interfere with the melting curve analysis or with the CT values obtained for target gene amplification of the respective serotypes. In the melting curve analysis of the NTCs, primer–dimer formation was indicated by a Tm below 75°C, which is lower than the Tm values of target gene products obtained with our assays.
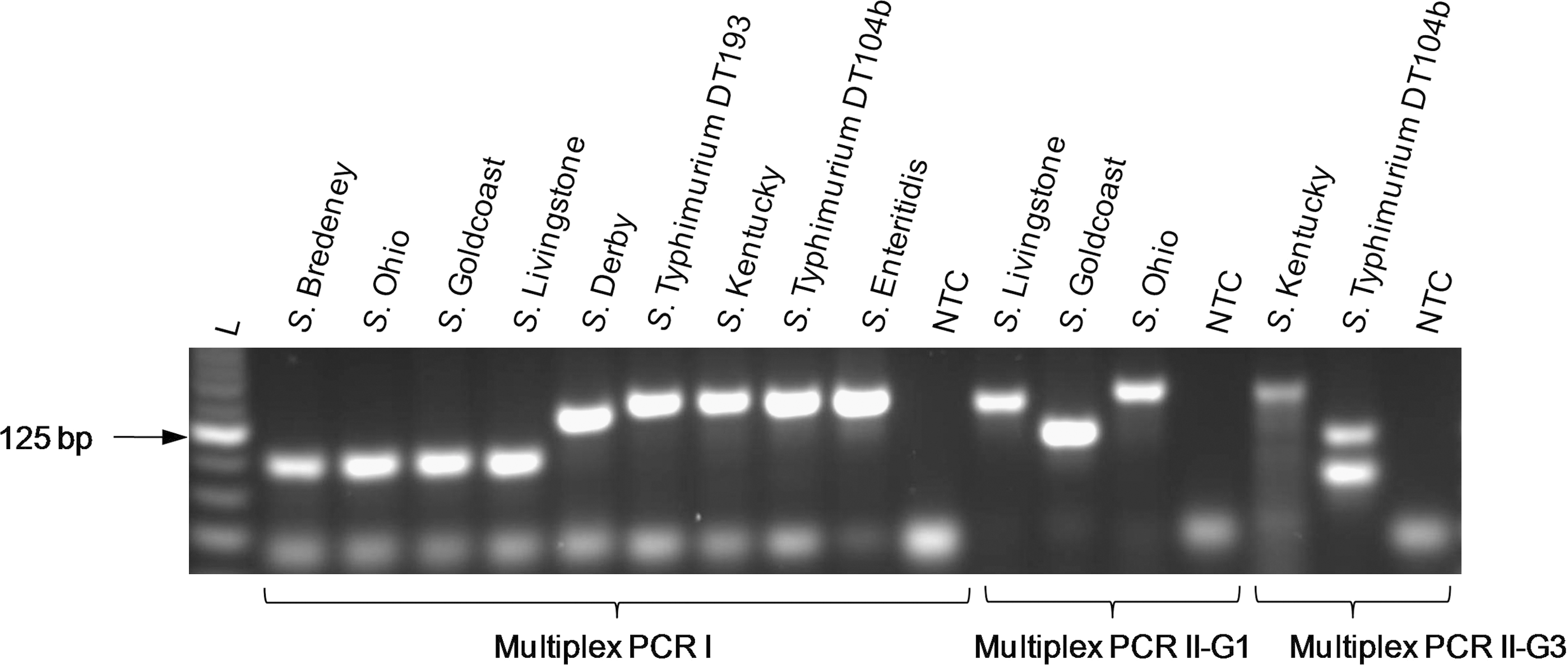
PCR products of Salmonella serotypes (used as initial test panel) generated by the Multiplex PCR I, Multiplex PCR II-G1, and Multiplex PCR II-G3 assays were run on a 3% agarose gel stained with GelRed (Biotium Inc.). L, 25 bp DNA Ladder; NTC, No template control.
Screening of the expanded test panel
The specificity of the established multiplex assays was verified on an expanded panel (97 strains) consisting of 85 Salmonella isolates (representing 37 serotypes) and on 12 non-Salmonella strains previously identified by phenotypic methods (Table 5). All Salmonella serotypes with the respective gene(s) targeted by the established Multiplex PCR assays were correctly identified as indicated by the positive CT values and the corresponding melting peak temperatures. Using the Multiplex PCR I assay 19 Salmonella serotypes produced positive results as determined by the CT values of their fluorescence curves and distinct melting temperatures of the melting peaks. Isolates that possessed the respective gene(s) included in the Multiplex PCR I reaction were classified into Groups 1–5, as established previously. Strains that were assigned into Group 1 and 3 required a second-step differentiation using the Multiplex PCR II-G1 or II-G3 assay, respectively. The established multiplex reactions correctly grouped and identified all strains tested, thus confirming previously obtained results.
Standard deviations of the mean values are indicated in parentheses.
Isolate(s) received from UCD Centre for Food Safety, Dublin, Ireland.
Isolate received from Backweston Laboratory Complex, Ireland.
Included S. Bredeney NCTC 05731 (received from UCD Centre for Food Safety, Dublin, Ireland).
Isolate(s) received from Laboratory of Food Microbiology and Infectious Diseases, UCD, Ireland.
Gene target is present, but Multiplex PCR II-G1 or II-G3 assay was not used for identification of this S. serotype.
Included S. Kentucky NCTC 05799 (received from UCD Centre for Food Safety, Dublin, Ireland).
Non-ACSSuT-type strains [included S. Typhimurium DT12 (2), U314 (1), and U340 (2)].
Included S. Typhimurium ATCC® 14028 (received from UCD Centre for Food Safety, Dublin, Ireland).
S. Typhimurium DT12 strains harbored the spv operon.
ACSSuT-type strains (included S. Typhimurium DT104 and DT104b); all strains harbored the spv operon as determined by additional screening using the Multiplex II-G3 assay.
Included S. Typhimurium NCTC 13348 (received from UCD Centre for Food Safety, Dublin, Ireland).
Included S. Enteritidis ATCC® 13076 (received from Laboratory of Food Microbiology and Infectious Diseases, UCD, Ireland).
NA, Not amplified by Multiplex PCR I, no subsequent screening by the Multiplex PCR II-G1 and II-G3 assays.
For full name and source see Table 2.
NTC, No template control (included NTC of Multiplex PCR I, II-G1, and II-G3, each in triplicate).
In addition to the Salmonella strains used in the initial test panel, the following serotypes were classified into one of the five groups based on the presence of the same target gene(s): Salmonella Give, Salmonella London, Salmonella Panama, and Salmonella Worthington were classified into Group 1; Salmonella Agona, Salmonella Berta, Salmonella Havana, Salmonella Rissen, and Salmonella Senftenberg were assigned to Group 2. Presence of the i-antigen allowed classification of the monophasic serotype 4,[5],12:i:- into Group 3. Salmonella Kedougou expresses a first-phase i-antigen as well as the second-phase antigens l,w, both targeted by primers FliC-i (Group 1) and FliC/FljB-l (Group 3) used in the Multiplex PCR I assay. The latter serotype displayed a distinctive melting curve pattern with two melting peaks. However, the observed melting peak produced by primer FliC/FljB-l was smaller than peaks generated by serotypes solely amplified by the same primer, most probably as a result of intercalation of SYBR Green I. Of the remaining isolates, 18 Salmonella serotypes and all 12 non-Salmonella strains produced negative results in all runs performed due to the lack of the respective flagellar antigen or unique gene sequence not included in the Multiplex PCR I assay. Salmonella serotypes which tested negative generated no target amplification. Negative results were indicated by amplification exceeding 26 cycles. Dissociation curve analysis showed that nonspecific amplification as a result of primer–dimer formation and other nonspecific product formations were clearly distinguishable from target amplification (Fig. 4). Their melting curve profiles were either similar to those of the negative control, with a Tm of <75°C, or displayed a small-shoulder melting peak with a Tm nonindicative for the target genes of interest.
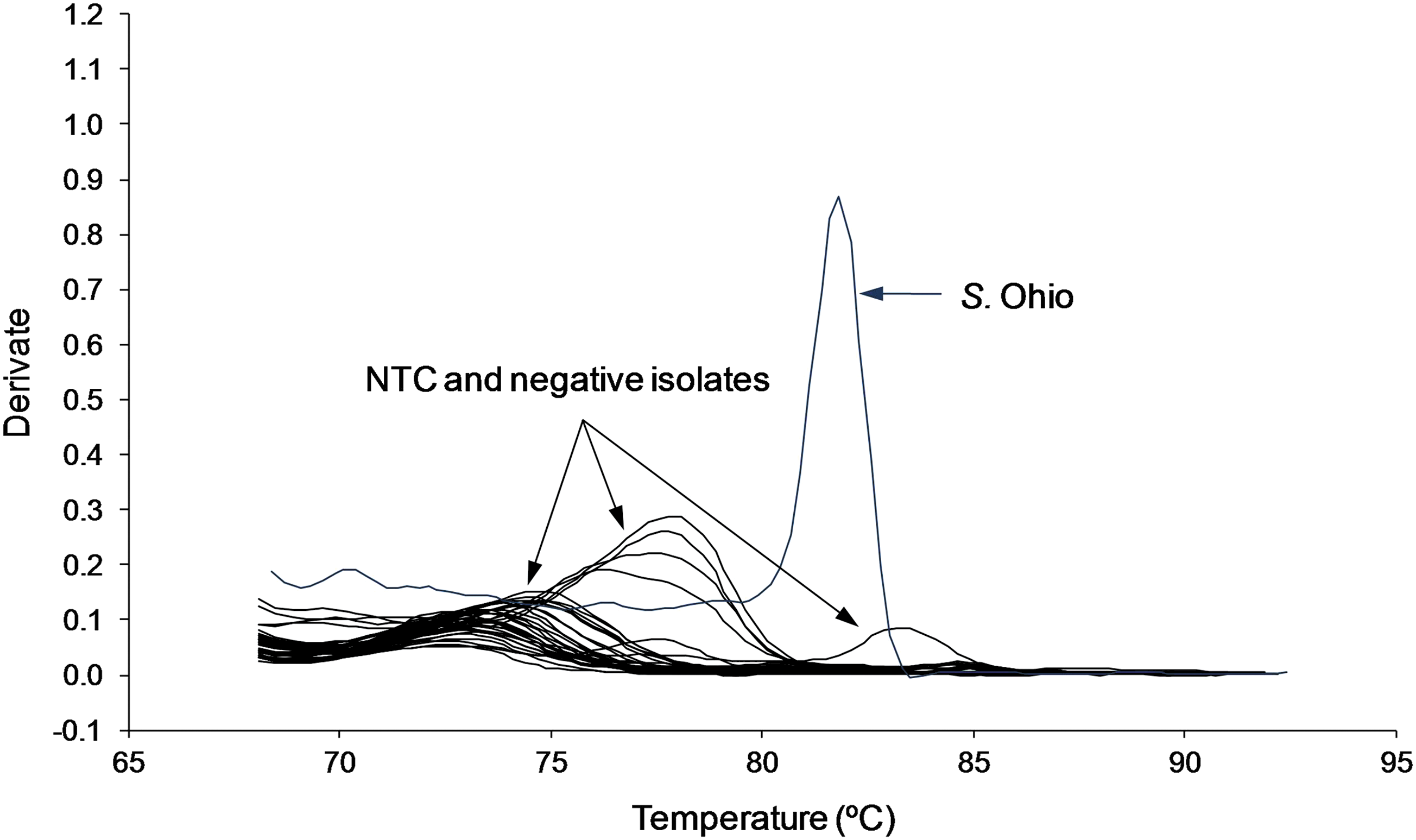
Melting curve pattern produced by the Multiplex PCR I assay for the NTC and negative isolates (Salmonella serotypes without target genes and non-Salmonella strains) compared to target amplification obtained for Salmonella Ohio.
Discussion
Rapid identification of Salmonella serotypes is crucial in monitoring and clinical outbreak investigations of this pathogen as it allows detection of epidemiological patterns and facilitates current surveillance systems as well as future prevention strategies. A two-step multiplex approach was chosen in this study for the development of a rapid, specific, and accurate real-time SYBR Green I PCR assay with melting curve analysis for identification of multiple Salmonella serotypes.
In the first step, a pentaplex real-time PCR assay (Multiplex PCR I) detected 19 prevalent Salmonella serotypes, designated into five groups. Certain serotypes assigned to Group 1 and serotypes in Group 3 were further identified by the second-step reactions Multiplex PCR II-G1 and II-G3, respectively.
The Multiplex PCR I assay allowed for accurate detection of Salmonella Bredeney, Salmonella Give, Salmonella London, Salmonella Panama, and Salmonella Worthington (Group 1); however, additional conventional serotyping is required for complete identification, as these serotypes were not targeted by the Multiplex PCR II-G1 assay. Similarly, Salmonella Agona, Salmonella Berta, Salmonella Derby, Salmonella Havana, Salmonella Rissen, and Salmonella Senftenberg (all classified into Group 2) could not be distinguished in a second-step reaction (on the basis of fljB genes) as these serotypes are monophasic. Hence, additional confirmatory tests are required to fully identify these serotypes. The multidrug-resistant (ACSSuT-type) strains of Salmonella Typhimurium (Group 4), Salmonella Enteritidis (Group 5), and Salmonella Kedougou (targeted simultaneously by Group 1 and 3 primers) were identified by their distinctive melting curve temperatures and required no further identification step. The SGI-1 island carrying the ACSSuT-resistance pattern as found in Salmonella Typhimurium DT104 and DT104b has previously been detected in related phage types such as DT120, U302, and DT12; however, phage types DT104 and DT104b currently constitute the vast majority of ACSSuT-type isolates within the EU (Anonymous, 2010; Perugini et al., 2010; Targant et al., 2010).
The Multiplex PCR II-G1 assay discriminated among Salmonella Goldcoast, Salmonella Livingstone, and Salmonella Ohio, whereas the Multiplex PCR II-G3 reaction distinguished between Salmonella Kentucky as well as non-ACSSUT-type strains of Salmonella Typhimurium. In addition, the latter assay was able to simultaneously detect the presence of Salmonella Typhimurium virulence plasmid frequently encountered in this serotype. Serotype-specific virulence plasmids as found in Salmonella Typhimurium, Salmonella Enteritidis, Salmonella Dublin, Salmonella Choleraesuis, Salmonella Gallinarum, Salmonella Pullorum, and Salmonella Abortusovis have been suggested to contribute to the expression of virulence and therefore to pathogenesis of these serotypes (Rychlík et al., 2006).
Primers were specifically designed to generate PCR amplicons of various sizes and distinctive GC contents. Fluorescence melting curve analysis of PCR products was used to discriminate between generated amplicons, as their Tm is a function of both the fragment length and the GC content (Ririe et al., 1997). This is also in agreement with our results, where high Tm values of PCR products were achieved by choosing targets of larger size or a high GC content. Although nonspecific amplification and primer dimer might occur, analysis of melting curves and CT values can easily differentiate between nonspecific and target amplification (Yoshitomi et al., 2006).
In summary, the established real-time multiplex SYBR Green I PCR assays successfully detected 19 Salmonella serotypes and further identified 7 serotypes, including Salmonella Ohio, Salmonella Goldcoast, Salmonella Livingstone, Salmonella Kedougou, Salmonella Enteritidis, Salmonella Kentucky, ACSSuT-type Salmonella Typhimurium DT104 and DT104b, as well as non-ACSSuT-type Salmonella Typhimurium strains. A particular advantage of our assays is their speed, saving time and labor, compared to individual PCR assays or phenotypic methods such as serotyping. The ability to quickly detect the presence of unexpressed flagellar genes is a major benefit compared to traditional methods. Further, the assays are easy to perform without the need of DNA extraction kits. Using this method, isolates can be screened and identified within the same day compared to traditional serotyping methods, which require several days due to subculturing and additional biochemical testing needed for identification of Salmonella serotypes (Anonymous, 2002). The multiplex PCR assays are particularly usefully when screening large numbers of Salmonella isolates. The high-throughput capability allows analysis of nearly 100 samples within ∼2 to 4 h. Although our method covers a broad range of the most frequently isolated Salmonella serotypes from both humans and animals in the EU, it is particularly useful for screening of porcine and bovine isolates as it can identify the majority of serotypes that currently predominate in these animals and associated meat products. The established assays cannot fully replace traditional serotyping, but their ability to quickly identify major serotypes makes them a fast and reliable alternative to such conventional approaches and complements current tools available for serotyping.
Conclusions
Our real-time multiplex SYBR Green I PCR assay is a rapid and specific method allowing for identification of multiple Salmonella serotypes frequently associated with humans, animals, and animal products within the EU. Its ability to simultaneously screen for the presence of the ACSSuT-type multidrug resistance pattern and virulence plasmid in Salmonella Typhimurium makes it a particularly attractive screening tool.
Footnotes
Acknowledgments
This research was funded by the Food Institutional Research Measure (FIRM) of the Department of Agriculture and Food under the National Development Plan, FIRM/06RDTAFRC471. The authors acknowledge the assistance of Dr. Annetta Zintl and Dr. Jorge Gutierrez Merino in the real-time PCR development and data analysis. We would also like to thank Dr. Montserrat Gutierrez and Sharon J. Duggan of the Central Veterinary Research Laboratory, Backweston, Celbridge, Co., Kildare, Ireland, who kindly provided some of the bacterial strains used in this study.
Disclosure Statement
No competing financial interests exist.