
Abstract
Campylobacters other than Campylobacter jejuni or C. coli were isolated in 35% of 343 farms recently analyzed in northern Spain. This study was aimed at identifying at the species level the 120 isolates collected (21 ovine, 52 beef cattle, 44 dairy cattle, and 3 porcine) by species-specific polymerase chain reaction and 16S rRNA gene sequencing analysis. Thus, five species were identified: Campylobacter hyointestinalis (90 isolates), Campylobacter lanienae (12), Campylobacter fetus subsp. fetus (10), Campylobacter lari (1), and Campylobacter sputorum (1). Ambiguous results were obtained for six isolates. Phylogenetic analyses of the 16S rRNA gene sequence placed three of them (cattle isolates) as an intermediate clade between C. hyointestinalis subsp. hyointestinalis and C. fetus, two ovine isolates formed a new clade clustering with Campylobacter concisus despite sharing higher similarity with Campylobacter mucosalis, and one porcine isolate shared similarly high homology with C. lanienae and C. hyointestinalis subsp. lawsonii. C. hyointestinalis was the predominant species, particularly in cattle, but it was also isolated from sheep and swine. C. lanienae was only found in sheep, C. fetus in cattle and sheep, and C. lari in a single dairy cattle farm. Although previously reported, the isolation of C. lari from cattle is not common, and this is the first report of C. lanienae and C. hyointestinalis in sheep. This study demonstrated the wide distribution in livestock of several emerging zoonotic Campylobacter species and provided valuable information on their host animal reservoirs.
Introduction
Here, to get a better insight into the occurrence of different Campylobacter spp. in livestock, we used previously described species-specific PCRs targeting the 16S and 23S rRNA genes to identify at the species level thermotolerant Campylobacter species other than C. jejuni and C. coli isolated from healthy ruminants and swine in farms from the Basque Country in northern Spain (Oporto et al., 2007). Further, 16S rRNA gene sequencing and phylogenetic analyses were carried out to confirm or resolve inconclusive PCR results.
Materials and Methods
Campylobacter isolates
In a prevalence survey of campylobacters in livestock carried out in the Basque Country (northern Spain) in 2003–2006 (Oporto et al., 2007), fecal samples were collected from 343 herds (124 beef cattle, 82 dairy cattle, 120 sheep, and 17 swine). Briefly, rectal fecal samples were diluted 1/10 for enrichment into Preston broth and incubated for 18–20 h at 42°C. The broths were then subcultured into Campylosel plates (bioMerieux) and incubated for 48–72 h microerobically (3% H2, 5% O2, 5% CO2, 87% N2). Thermotolerant campylobacters were isolated from 203 herds, identifying C. jejuni and C. coli in 20.7% and 6.4% of the herds, respectively. However, in 120 (21 sheep, 3 swine, 52 beef cattle, and 44 dairy cattle) of them, isolates positive for a Campylobacer genus-specific PCR but negative to C. coli- and C. jejuni-specific amplification were then classified as other thermophilic campylobacters (OTCs) (Oporto et al., 2007). These 120 isolates were the target of the present study.
Species identification by PCR
A multiplex PCR using primers described elsewhere (Bastyns et al., 1994; Logan et al., 2000; Inglis and Kalischuk, 2003) was developed to identify Campylobacter hyointestinalis, Campylobacter fetus, and Campylobacter lanienae. A second PCR targeting the 16S rRNA gene as described by Linton et al. (1996) was used to identify Campylobacter lari. Sequence of each primer (Sigma-Genosys), concentration used, and estimated product size are shown in Table 1. All amplification reactions consisted of 25 μL mixtures containing 20 ng of genomic DNA, 1×PCR buffer (200 mM Tris-HCl [pH 8.4] and 500 mM KCl), 0.2 mM deoxynucleoside triphosphates, 2 mM MgCl2 for multiplex PCR and 1.5 mM for C. lari PCR amplification, and 1 U of Taq DNA polymerase (Invitrogen). Cycling conditions for the multiplex PCR were as follows: 4 min at 95°C; 30 cycles of 30 sec at 94°C, 30 sec at 55°C, and 60 sec at 72°C; and a final extension step of 5 min, with annealing at 57°C for C. lari PCR amplification. In addition, a further multiplex PCR was performed to confirm C. fetus isolates identification and to determine the subspecies as previously described by Hum et al. (1997).
PCR, polymerase chain reaction.
16S rRNA gene sequencing analysis
The complete 16S rRNA gene was amplified using primers described by Lawson et al. (1998). Purified PCR amplicons were partially (550–900 nt) or completely (∼1500 nt) sequenced using the Big Dye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems) and primers described in Table 1. The sequenced products were analyzed with an ABI3130 automated sequencer (Applied Biosystems). All sequences were compared with the NCBI GenBank nonredundant nucleotide database using BLASTN and aligned along with data retrieved from GenBank with ClustalW using the Vector NTI suite 8.0 software (InforMax). A gap opening penalty of 15 and a gap extension penalty of 6.66 were used in the multiple alignments. Intervening sequences (IVSs) were manually deleted from the 16S rRNA gene sequences for phylogenetic analysis. Similarity matrices were constructed from aligned sequence data by single distance using the Jukes and Cantor (Jukes and Cantor, 1969) or the Kimura's two-parameter model (Kimura, 1980). Phylogenetic trees were constructed by the neighbor-joining (Saitou and Nei, 1987) and the maximum parsimony methods as implemented in the Mega 4 package (Kumar et al., 2001), deleting all gap sites and including Sulfurospirillum barnesii and Sulfurospirillum deleyianum as outgroup. Stability or accuracy of inferred topologies was assessed via bootstrap analysis (Felsenstein, 1985) of 1000 replicates. The nearly complete 16S rRNA gene sequences determined in this study were submitted to GenBank under accession numbers HQ628642–HQ628649.
Results
PCR produced a single band of the expected sizes from all except four isolates that were negative for all species-specific PCR amplifications (Table 2). All the isolates identified as C. fetus by 23S rRNA gene-based PCR were confirmed as C. fetus and identified as subspecies fetus by the amplification of the carbon starvation protein A and lack of amplification with the venerealis subspecies-specific primers. Sequence analysis was performed to confirm or resolve inconclusive PCR results. Thus, the complete 16S rRNA gene was sequenced for the four isolates negative by species-specific PCR (C0102-DC, C0078-Sw, C0128-Sh, and C0162-Sh isolates), whenever partial sequencing resulted in ambiguous identification (C0027-BC, C0066-DC and C0002-Sw isolates) and when the identified Campylobacter species had been isolated from hosts not previously reported (C0292-Sh and C0098-Sh isolates) (Table 3). Further, partial 16S rRNA gene sequencing confirmed PCR results in a selection of 33 isolates. A summary of the species identification of the 120 isolates and their distribution by animal source resulting from these analyses are shown in Tables 2 and 3. Briefly, 90 isolates were identified as C. hyointestinalis, 12 as C. lanienae, 10 as C. fetus subsp. fetus, 1 as C. lari, and 1 as Campylobacter sputorum. Ambiguous results were obtained for the remaining six isolates (C0102-DC, C0078-Sw, C0128-Sh, C0162-Sh, C0027-BC, C0066-DC, C0281-BC), because phylogenetic analysis of their complete 16S rRNA gene placed them as outliers to defined groups, but with relatively low bootstrap support (Fig. 1) and, sometimes, in contradiction with PCR results (Table 3).
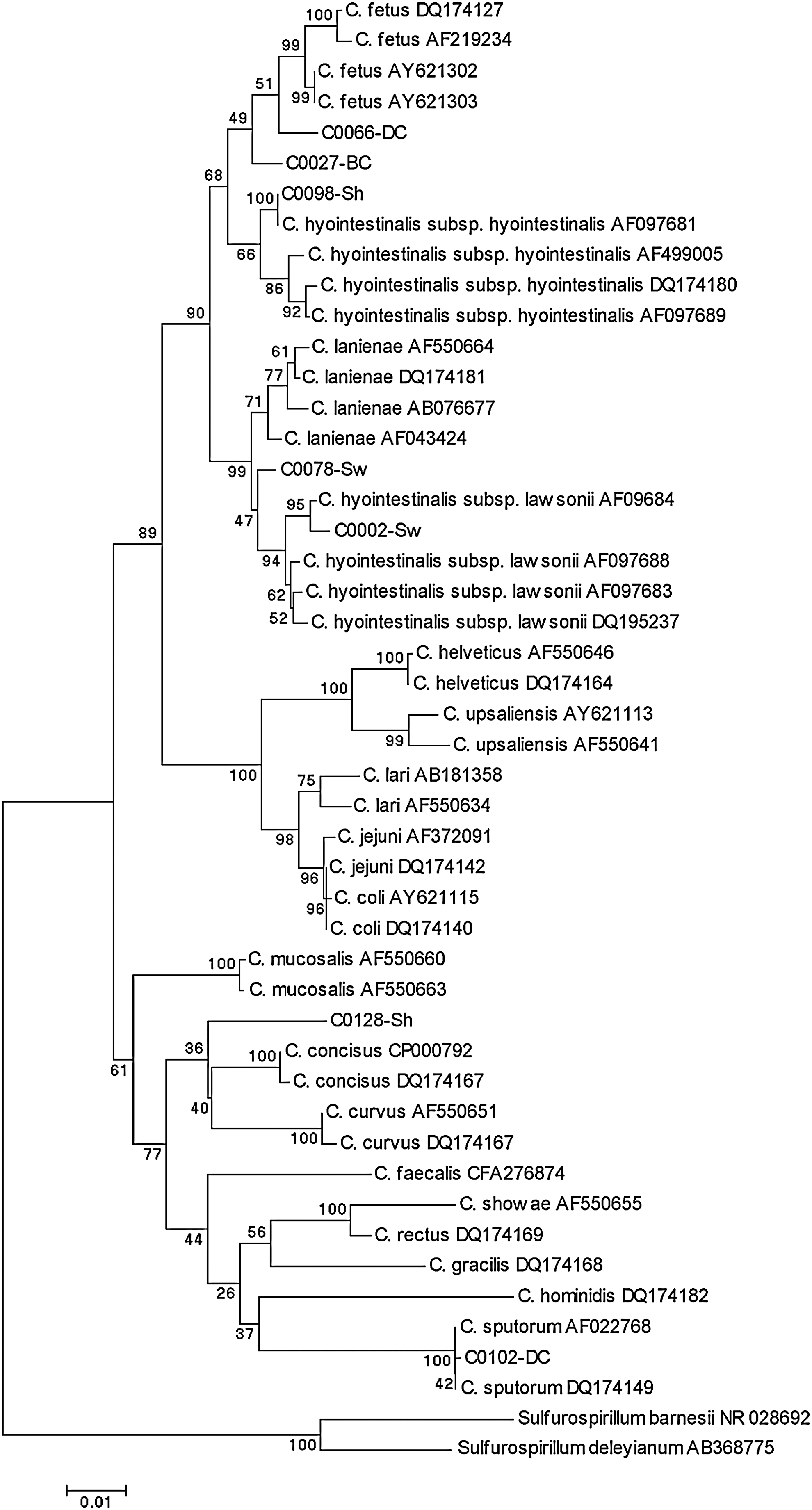
Phylogenetic tree comparing 1289 sites of the 16S rRNA gene of the campylobacters sequenced in this study and the main Campylobacter species retrieved from GenBank. A gap opening penalty of 15 and a gap extension penalty of 6.66 were used in the multiple alignments with Vector NTI. Distances were estimated by Kimura's two-parameter model and the tree was constructed by neighbor-joining using the MEGA 4.1 package. The percentage of 1000 bootstrap replicates supporting each branch is indicated at the nodes. Scale bar used was nucleotide substitutions per position.
Four isolates (C0128-Sh, C0162-Sh, C0078-Sw, and C0102-DC) negative for all species-specific PCRs (see Table 3 and Fig. 1 for more details on complete 16S rRNA sequencing results).
Three isolates (C0027-BC, C0281-BC, and C0066-DC) positive for C. hyointestinalis-specific PCR and negative for both C. fetus-specific PCRs; high similarity was observed in their 16S rRNA gene to both C. hyointestinalis subsp. hyointestinalis and C. fetus, and phylogenetic analysis placed them as an intermediate cluster (see Table 3 and Fig. 1 for sequencing results).
Sh, sheep; Sw, swine; BC, beef cattle; DC, dairy cattle.
Total size including the IVS.
Isolate C0128-Sh was 100% identical to C0162-Sh in the 1396 nt sequenced of the latter.
A homology of 98.8% between both cattle isolates (17/1442 different positions); C0066-DC was identical to another bovine isolate (C0281-BC, 700 nt sequenced).
Identical to three other isolates from beef cattle partially sequenced (528–910 nt).
IVS, intervening sequence.
Partial sequencing confirmed the identity of the C. lari isolate from dairy cattle (identical to AF550634 and DQ17414), C. fetus from sheep and beef cattle (identical between them and to AY621110), and the 12 C. lanienae from sheep (also identical). The nearly complete gene was sequenced for one C. lanienae isolate from sheep (C0292-Sh) and showed 99.8% similarity with bovine C. lanienae AY288304 (Table 3). More diversity was found among the C. hyointestinalis isolates sequenced, in which both subspecies hyointestinalis and lawsonii were identified. Nineteen isolates were identified as C. hyointestinalis subsp. hyointestinalis (16 from cattle, 2 from sheep, and 1 from swine) and had five different sequences with similarities among them and with other sequences of C. hyointestinalis subsp. hyointestinalis from the database of 98.9%–100%. In particular, the nearly complete 16S rRNA gene sequenced from an ovine isolate (C0098-Sh) was identical to C. hyointestinalis subsp. hyointestinalis AF097681 (Table 3 and Fig. 1). C. hyointestinalis subsp. lawsonii was represented by a porcine isolate (C0002-Sw) that shared 99.1% similarity with C. hyointestinalis subsp. lawsonii AF097688 (Table 3) and clearly clustered with this subspecies (Fig. 1).
Enlarged 16S rRNA gene amplicons (∼1725-bp as opposed to the expected 1495-bp fragment) were obtained with primers CG12F and CG1507R for C. sputorum C0102-DC and C. hyointestinalis subsp. lawsonii C0002-Sw. Sequence analysis revealed the presence of IVS inserted following base position 185 of the C. jejuni NCTC 11351T (AF372091) 16S rDNA, where a deletion of 3 nt was followed by IVSs sized 228 and 230 bp, respectively, but with very different sequences (72.2% similarity). Blast searches identified the IVS within C. lanienae AB076676 as the closest hit to the IVS in C0002-Sw (217/230 bp identical). Also, C. hyointestinalis subsp. lawsonii AF097684 IVS was similar, but it was larger and had a 15-bp insertion. The IVS in C0102-DC was nearly identical (98.3%, 227/231) to the IVS in C. sputorum AB501345.
Discussion
Molecular techniques, frequently based on the rRNA genes, are of invaluable help in detecting, identifying, and determining phylogenetic relationships among different organisms. Here, PCR identification of several Campylobacter species was based on the specific amplification of different regions of the small and large subunit rRNA genes using primers described elsewhere (Bastyns et al., 1994; Linton et al., 1996; Logan et al., 2000; Inglis and Kalischuk, 2003). Results were confirmed by partial and, in some instances, complete 16S rRNA gene sequencing and phylogenetic analyses. Bootstrap support was high for nodes that could be unambiguously resolved by the different tree reconstruction methods, suggesting a coherent branching order. This was the case for porcine isolate C0002-Sw, clearly identified as C. hyointestinalis subsp. lawsonii, and C0102-DC, negative for all species-specific PCRs and identified as C. sputorum by sequencing analysis.
However, in some cases, phylogenetic analysis of the complete gene provided inconclusive results, placing some isolates as outliers to defined groups with relatively low bootstrap values. In some instances, this was due to the low levels of 16S rRNA gene variations between the species C. hyointestinalis subsp. lawsonii and C. lanienae (minimum diversity, 1.9%), and C. hyointestinalis subsp. hyointestinalis and C. fetus (minimum diversity, 1.6%) (Gorkiewicz et al., 2003). Thus, bovine isolates C0027-BC and C0066-DC, positive for C. hyointestinalis-specific PCR and negative for both C. fetus-specific PCRs, formed intermediate branches between C. hyointestinalis subsp. hyointestinalis and C. fetus in the phylogenetic tree. Other primers described for the specific identification of C. fetus based on the 16S rRNA gene (Linton et al., 1996) would not amplify these strains because of a mismatch in the 3′ end of the reverse primer, suggesting that these two isolates could be presumptively classified as C. hyointestinalis subsp. hyointestinalis. Porcine isolate C0078-Sw was negative for the four species-specific PCRs and clustered (47% bootstrap support) with C. hyointestinalis subsp. lawsonii, but showed higher homology with C. lanienae. Sequence analysis of this and several other C. lanienae sequences from GenBank revealed three polymorphic sites within the annealing site for the C. lanienae reverse primer, which could explain amplification failure. 16S rRNA gene sequence data for this sample were therefore most consistent with the identification of C0078-Sw as C. lanienae.
Species identification by 16S rRNA gene sequencing analysis was, however, uncertain for the ovine isolates C0128-Sh and C0162-Sh, also negative for the species-specific PCRs. Both isolates were identical in their 16S rRNA gene sequence and formed a new branch that clustered with Campylobacter concisus despite sharing higher similarity with Campylobacter mucosalis. C. mucosalis has been isolated from pigs with porcine necrotic enteritis and ileitis, but has not been associated with human disease. Human infections initially attributed to C. mucosalis were later shown to be misidentified C. concisus infections (On, 1994), as both species are phenotypically similar. C. concisus is found in the human oral cavity and has been identified in stool and blood samples of children and adults with and without diarrhea, but its role as a human enteric pathogen remains unclear (Van Etterijck et al., 1996; Engberg et al., 2000). Similarly, this species has no known animal hosts. However, high levels of C. concisus shedding in diarrheic pet dogs have been recently reported (Chaban et al., 2010), and here, an isolate closely related to C. concisus was recovered from healthy sheep feces. The diversity of C. concisus isolates at the phenotypic and genotypic levels (Vandamme et al., 1989; Aabenhus et al., 2005; Engberg et al., 2005) and its particular isolation requirements might have hampered a deeper knowledge regarding their animal hosts. Here, the investigation of other genes or a combination of different phenotypic and genotypic profiles using well-established methods in a polyphasic approach (On, 2001) would be needed to identify these two ovine isolates or verify whether they represent a new taxon.
Although isolation rates observed in this study could be biased by the isolation conditions used and should therefore not be considered an estimation of the true prevalence of the different species identified, recovery rates for Campylobacter species other than C. jejuni and C. coli in healthy ruminants and swine were relatively high. According to PCR and sequencing results, the most predominant species in farms from the Basque Country was C. hyointestinalis, and it was present in the four production systems. The second was C. lanienae, only found in sheep herds, and closely followed by C. fetus, which was isolated from cattle and sheep. Finally, the only C. lari and C. sputorum isolates detected were found in dairy cattle. Putting these results in the frame of the prevalence study where these isolates were obtained (Oporto et al., 2007), the high proportion of herds positive for OTC in the Basque Country (120/343) was mainly associated with bovine herds infected with C. hyointestinalis. In fact, C. hyointestinalis isolation rate (26.2%) was above that estimated for C. jejuni (20.7%) and C. coli (6.4%) (Oporto et al., 2007). C. hyointestinalis is commonly associated with cattle, but prevalences reported vary from values similar to those observed here (25.9%–32.0%) (Atabay and Corry, 1998; Busato et al., 1999) to much lower rates (8.0%–10.8%) (Inglis et al., 2003; Hakkinen et al., 2007). The sequence analyses performed here identified all of the cattle C. hyointestinalis sequenced as subspecies hyointestinalis. In swine, one isolate was identified as C. hyointestinalis subsp. hyointestinalis, the organism most frequently isolated from pigs with proliferative enteritis (Gebhart et al., 1985), and another as C. hyointestinalis subsp. lawsonii, which is commonly isolated from the stomach of healthy pigs. C. hyointestinalis of the subspecies hyointestinalis, as confirmed by sequencing analysis, was also isolated from sheep. As far as we are concerned, this is the first report of C. hyointestinalis detection in ovine feces. C. hyointestinalis subsp. hyointestinalis has also been isolated in human diarrheal cases (Lastovica and Allos, 2008) and its transmission from a pig to a human has been reported (Gorkiewicz et al., 2002).
C. lanienae was also detected in sheep for the first time in this study and the sequences shared similarity up to 99.8% with C. lanienae (AY288304) isolated from dairy cattle in Canada (Inglis and Kalischuk, 2003). This species was discovered during a routine screen of healthy abattoir workers (Logan et al., 2000), and so far, it has not been associated with human disease. It usually appears in the gastrointestinal tract of food animals such as cattle and swine, although debate exists regarding the main reservoir (Sasaki et al., 2003; Inglis et al., 2004; Guevremont et al., 2008). Guevremont et al. (2008) reported a 5 nt difference between C. lanienae strains from pig and dairy cattle in the 5′ region of the 16S rRNA gene sequence. Here, sheep isolate C0292-Sh shared its pattern with the bovine sequences. Therefore, it is important to characterize more C. lanienae strains to expand the knowledge on the genetic diversity of this species and its association with different animal reservoirs as well as its role in human gastroenteritis.
Interestingly, C. fetus was recovered from both sheep and cattle despite isolation at 42°C. Although thermotolerant C. fetus strains have been described, most strains grow better at 25°C and 37°C. More precisely, certain strains of C. fetus subsp. fetus but not C. fetus subsp. venerealis can grow at 42°C (van Bergen et al., 2008). Therefore, as expected, the 10 C. fetus isolates recovered in this study were confirmed as C. fetus subsp. fetus by PCR. C. fetus subsp. venerealis is only rarely reported to cause human disease, whereas C. fetus subsp. fetus has been found in cases of human diarrhea, septicemia, and abortion (Lastovica and Allos, 2008). In livestock, particularly sheep and cattle, both subspecies are the cause of important economic losses associated with infertility problems and abortion.
Although the best-known reservoir of C. lari includes various birds, isolation of C. lari from cattle, albeit previously reported, is not common (Aarestrup et al., 1997; Brown et al., 2004; Gilpin et al., 2009). C. lari can cause gastroenteritis and septicemia in humans and has been found in episodes of gastroenteritis in some birds, but no reports are available on any disease in cattle. Finally, C. sputorum is found in the oral cavity, feces, and skin lesions of humans, and strains have also been isolated from the feces of sheep and cattle, but pathogenicity for neither humans nor animals is known (On et al., 1998).
The presence of IVSs in the 16S rRNA gene of Campylobacter spp. was first described by Linton et al. (1994), who reported that certain strains of Campylobacter helveticus contained an IVS of ∼150 bp. The IVSs reported here add to the list of different IVSs that have been reported since then in the 16S rRNA gene of several Campylobacter species, including C. sputorum and C. hyointestinalis (Etoh et al., 1998; Harrington and On, 1999).
Conclusions
This study demonstrated the wide distribution of several thermotolerant Campylobacter species other than C. jejuni and C. coli in farm animals. C. lanienae and C. hyointestinalis were isolated from sheep for the first time, adding to the knowledge on these organisms and their potential reservoir hosts, which may act as possible origin of bacterial contamination of food products. Analysis of the complete 16S rRNA gene revealed a high degree of sequence diversity, which, in some cases, precluded clear species identification, and this is of special concern for primer design and PCR-based detection procedures. Still, molecular detection and characterization of rarely occurring Campylobacter species are crucial to investigate their possible role in human diseases. Until their pathogenic potential is clarified, studies on the detection, identification, and prevalence of such taxa in the food chain would provide valuable epidemiological data.
Footnotes
Acknowledgments
This work was funded by the Basque Government (Departamento de Medio Ambiente, Planificación Territorial, Agricultura y Pesca), Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria INIA (RTA2006-00105-00-00), and FEDER.
Disclosure Statement
No competing financial interests exist.