
Abstract
The capability of forming biofilm makes the foodborne pathogen Listeria monocytogenes more resistant to environmental stresses. To examine the mechanism of biofilm formation, L. monocytogenes transposon mutant strain GB8 with decreased ability to form biofilm was characterized in this study. Southern blot assay revealed a single copy of transposon in the GB8 chromosome, and the insertion site was identified by inverse polymerase chain reaction (IPCR). The expression of lm.G_1497 was found to be inactivated by the transposon insertion, and the gene was identical to gene LMOf2365_1497 in the sequenced strain L. monocytogenes strain 4b F2365, encoding a MerR family protein. The ability of producing biofilm was recovered in revertant GBR8. It was confirmed by quantitative real-time PCR that the transcription of the gene flanking lm.G_1497 was not affected by Tn917 insertion. These results suggested that lm.G_1497 was responsible for the positive regulation of biofilm formation in L. monocytogenes 4b G. Moreover, the failure of lm.G_1497 expression resulted in a decrease of the autolysis rate, while no effect on cell growth and motility in L. monocytogenes GB8, which might imply that the reduction in biofilm was caused by the difference in cell autolysis.
Introduction
L. monocytogenes readily forms biofilms in food processing environments (Beresford, 2001; Gandhi, 2007). Biofilm is a complex mixture of bacterial cells, extracellular polymeric substances (EPS), water, and nutrients on a solid substrate (Costerton et al., 1999; Hall-Stoodley et al., 2004). L. monocytogenes in a biofilm shows a higher resistance to stress conditions than planktonic cells (Chen and Stewart, 2000; Holah et al., 2002). This characteristic makes the L. monocytogenes in biofilm difficult to remove by physical cleaning or chemical disinfecting in food processing environments. Therefore, the understanding of the mechanism of biofilm formation can contribute to the elimination of L. monocytogenes. It is believed that the biofilm cells of L. monocytogenes always confront many stress factors, such as limited nutrition, oxidative stress, high osmotic pressure, and increased toxic metabolites (Helloin et al., 2003; Takhistov and George, 2004). Under these circumstances, biofilm cells live with different molecular mechanisms from that of planktonic cells. Some stress-related genes have shown important roles in biofilm development. For instance, the sigB gene of L. monocytogenes, coding for the regulator of stress response genes, is activated in biofilm (Van der Veen and Abee, 2010). The oxidative stress response gene sod is also proven to be up-regulated in L. monocytogenes biofilm (Tremoulet et al., 2002). Unfortunately, it is still not clear about the mechanism of biofilm formation in L. monocytogenes.
Recently, the mutant GB8 with decreasing capability of forming biofilm was isolated by screening a library constructed via transposon Tn917 mutagenesis of L. monocytogenes 4b G (serotype 4b) (Ma and Shi, 2009). In the present study, the inactivated gene of GB8 was identified, and its function was elucidated for better understanding the biofilm formation in L. monocytogenes.
Methods
Bacterial strains
L. monocytogenes 4b G was obtained from Chinese Center for Disease Control, and Tn917 insertion mutant GB8 was constructed in our laboratory (Ma and Shi, 2009). Escherichia coli DH5α containing plasmid pKSV7 used for constructing revertant was provided by Dr. Sarah Vela (University of California at Berkeley). L. monocytogenes was routinely cultured in brain-heart infusion (BHI) broth (BD, Franklin Lakes, NJ) and E. coli in Luria-Bertani (LB) broth (Oxoid, Cambridge, UK). Antibiotics (Sigma, Mississauga, Canada) were supplemented as follows: 5 μg/mL of erythromycin (erm) for GB8, 50 μg/mL of ampicillin (amp), and 10 μg/mL of chloramphenicol (cm) for E. coli DH5α containing plasmid pKSV7. The stock cultures were stored at −80°C in 20% glycerol (Table 1).
Southern blot analysis
The insertion of transposon Tn917 into the chromosome of wild-type L. monocytogenes 4b G was confirmed by Southern blot analysis. Genomic DNA was digested with EcoRI and transferred onto a Hybond-N+ nylon membrane (GE Healthcare, Waukesha, WI). The 525-bp Tn917 fragment amplified with primers Tn4292 and Tn3768 (Table 2) was used as a probe for hybridization. The probe labeling and southern hybridization were performed with the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche Applied Science, Indianapolis, IN). The hybridization signals were detected after exposing to x-ray film for 25 min.
Location in the published nucleotide sequence on Tn917 (accession number M11180).
Location in the published complete genome on L. monocytogenes serotype 4b strain F2365 (accession number AE017262).
PCR, polymerase chain reaction; IPCR, inverse polymerase chain reaction.
Identification of the Tn917 insertion site in GB8
Inverse PCR (IPCR) was performed to identify the flanking regions adjacent to the Tn917 transposon (Ochman et al., 1990). Chromosomal DNA of GB8 was digested with HindIII and self-ligated with T4 DNA Ligase, as the template for IPCR. Primers Tn365 and Tn2485 were designed to clone the Tn917 5′ flanking sequence, while primers Tn4431 and Tn5054 were used to obtain the 3′ flanking sequence. For PCR, each 25-μL reaction volume contained the following: 2.5 μL of 10×PCR buffer, 1.0 μL of deoxyribonucleotide triphosphate (dNTP), 1.5 μL of MgCl2, 1.0 μL of each 10 mM stock primer pair, 2 μL of template DNA, and 0.2 μL of EX Taq. PCR cycle parameters were as follows: an initial denaturation step at 95°C for 5 min; 34 cycles of 94°C for 30 s, followed by 50°C for 30 s, and 72°C for 2 min; and a final extension step at 72°C for 8 min. DNA sequences of the PCR products were determined (Sangon Co. Ltd., Shanghai, China) and aligned using BLASTn online software (
Construction of the revertant GBR8
The construction of the revertant GBR8 was performed as described by Zhu et al. (2008). In brief, a 1040-bp DNA fragment containing the Tn917 insertion site was prepared by PCR from the wild-type chromosomal DNA using primers 8IF and 8IR. This fragment contained a predicted open reading frame identified by ORF Finder (
Reverse transcription PCR
Successful homologous recombination of lm.G_1497 was confirmed by reverse transcription PCR with primers 8F and 8R. RNA of Listeria cells was extracted with Takara Total RNA Isolation kit (Takara, Otsu, Shiga, Japan), and cDNA was synthesized by reverse transcription with Takara kit DRR 047A (Takara) according to the directions given by the supplier.
For analysis of lm.G_1496 expression, quantitative real-time PCR assay was performed using primers 1496F and 1496R. Three samples of each cDNA preparation were amplified for one cycle at 95°C for 5 min, 35 cycles with conditions of 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. 16S rRNA gene was used as the internal standards (Table 2).
Biofilm assay
Biofilm formation of L. monocytogenes was monitored using the microtiter plate assay described by Djordjevic et al. (2002). Briefly, L. monocytogenes was incubated in 96-well microtiter plates at 37°C for 72 h in eight replicates. After gently removing planktonic cells, 1.0% crystal violet was added to stain the adhered cells, and the unbound dye was removed by washing with sterile distilled water. The crystal violet bound to sessile cells in biofilm was solubilized with 96% ethanol, and the amount of biofilm was determined by measuring the optical density at 590 nm (OD590). The assay was repeated three times.
Triton X-100–induced autolysis assay
Autolysis assay was performed as described by Mani et al. (1993) with modifications. The wild-type GB8 and GBR8 were cultured in BHI broth to an OD600 of 0.7±0.05 in triplicate, following with cell collection by centrifugation (7000 rpm, 10 min, 4°C) and resuspended in the same volume containing 0.05M Tris-HCl (pH 7.2) and 0.05% Triton X-100. The cells were then incubated at 30°C with shaking, and the OD600 value was measured at 1-h intervals for 5 h. The percentage of culture density remaining was calculated by dividing the measured OD600 by the initial OD600×100%.
Growth curve analyses of Tn917 insertion mutants
The overnight cultures of the wild-type 4b G and mutants GB8, GBR8 of L. monocytogenes were diluted 1:100 in BHI broth, followed with incubation at 37°C to OD600=0.3. Then each strain was diluted at 1:50 (V/V) in BHI broth, and 200 μL of the final culture from each strain was added into the microtiter plates. The growth curve was measured by Labsystems Bioscreen C equipment (Rodovre, Denmark), and the OD600 was detected at 1-h intervals for 24 h at 37°C with shaking. Three replicates of growth curve determination for each strain were carried out in one single bioscreen.
Cell motility assay
Bacterial motility was tested on semi-solid agar plates as described by Knudsen et al. (2004) in triplicate. In brief, a single colony of the wild-type or mutant strain was inoculated in BHI solidified with 0.25% agar and incubated at 25°C and 37°C for 48 h, respectively.
Results
Identification of the Tn917 insertion site
To identify the insertion site of transposon Tn917, IPCR was performed with specific primers designed according to Tn917 sequence. The nucleotide sequencing of 5′ flanking related fragment of Tn917 transposon insertion showed that a 1.0-kb fragment of the IPCR product was comprised of part of Tn917 flanking sequences and a 458-bp fragment which sequence was 100% matched with LMOf2365_1497 in sequenced L. monocytogenes strain 4b F2365. Because of its homology, the inactivated gene was named lm.G_1497, and the gene product was a predicted MerR family transcriptional regulator. The insertion site of Tn917 was located at 15 bp before the ATG start codon of lm.G_1497. According to the complete sequenced L. monocytogenes strain 4b F2365, the transposon Tn917 was oriented with the same transcriptional direction of lm.G_1497, while the flanking genes of lm.G_1497 were opposite to lm.G_1497 (Fig. 1). Furthermore, the expression of lm.G_1496 in GB8 was the same as that in the wild-type L. monocytogenes 4b G (Fig. 2).
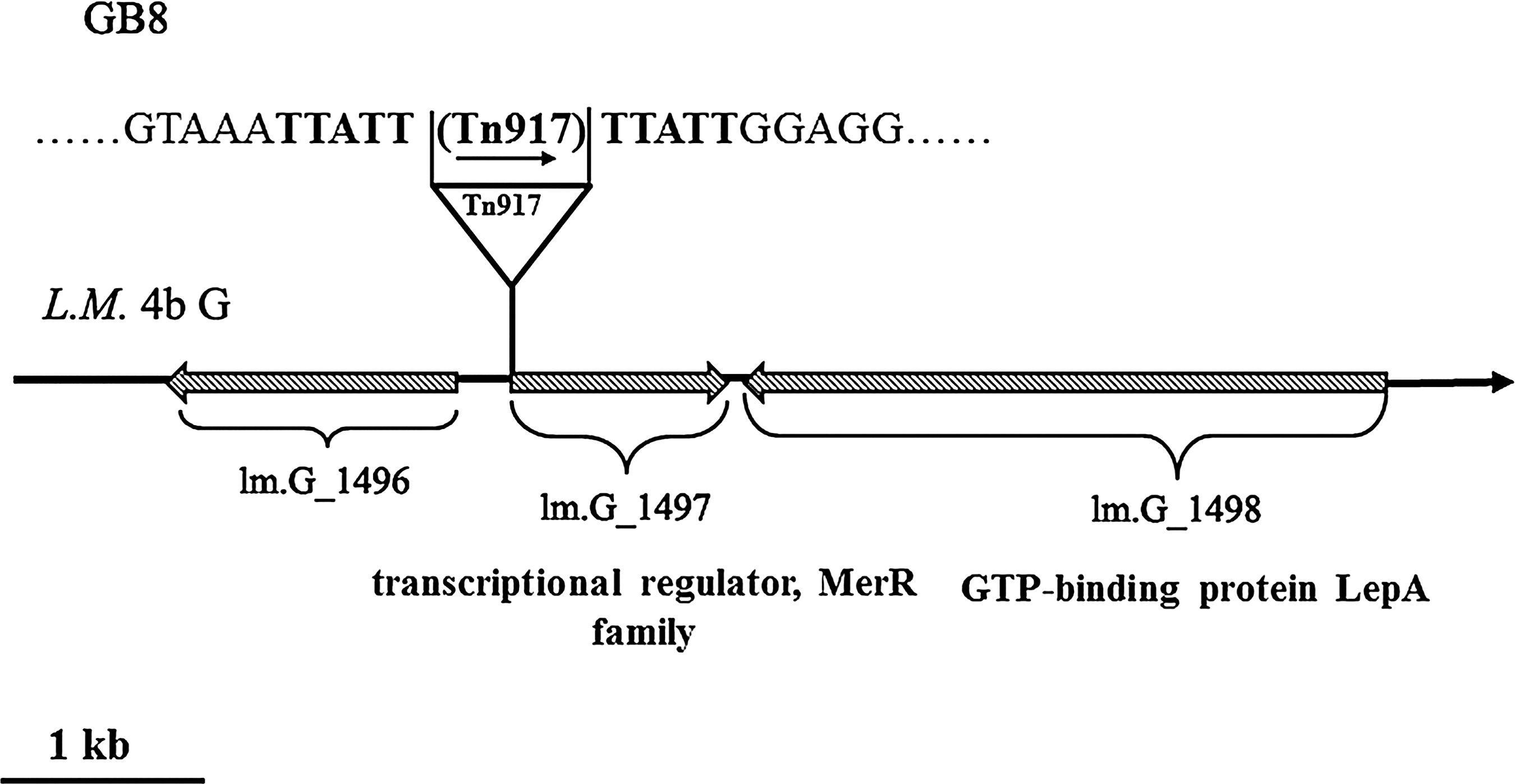
The insertion site and direction of Tn917 in Listeria monocytogenes GB8. The 3′ flanking sequence of Tn917 insertion site were confirmed by polymerase chain reaction (PCR) with a primer set (FA-F and FA-R) (Table 2). Arrows showed the direction of transcription, and the typical 5-bp target sequence duplicated by Tn917 transposition is indicated in boldface letters.
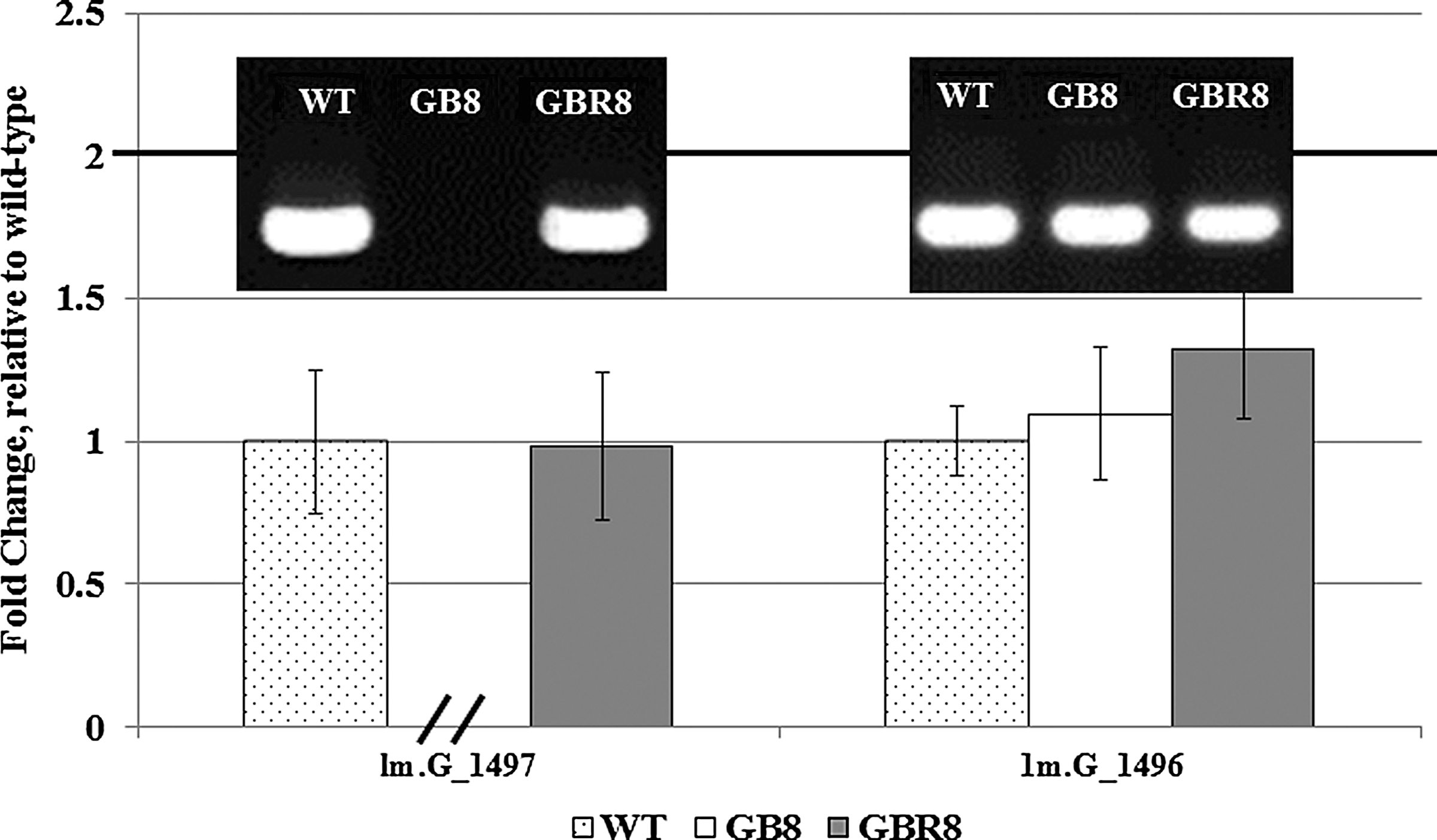
Confirmation for the effect of Tn917 insertion on the expression of lm.G_1497 and its flanking gene lm.G_1496 by quantitative real-time polymerase chain reaction (PCR). The two genes in Listeria monocytogenes GB8 (Tn917 insertion mutant) and L. monocytogenes GBR8 (revertant) were compared at the transcript levels with L. monocytogenes 4b G (WT [wild-type]). Agarose gel electrophoresis of quantitative real-time PCR products are shown on each column. The parallel (//) indicates no quantitative real-time PCR product of lm.G_1497 detected in GB8. This experiment was repeated three times, and Tukey's test result (p<0.05) verified that Tn917 insertion did not affect lm.G_1496 expression.
After the homology analysis of lm.G_1497 using the GenBank database, it was revealed that this MerR family regulator coding gene was highly conserved (more than 90% similarity in most of the sequenced Listeria strains) in Listeria species, and the production of Lm.G_1497 was also proven to be conserved in Listeria strains by protein blast. Bioinformatics analysis revealed that the lm.G_1497 encoding protein contained 145 amino acid subunits, which was the most homologous to the MerR family regulator in Bacillus species (51% identities), and the region from the 3rd amino acid subunit to the 114th amino acid subunit was homologous to a superoxide response regulon SoxR (Mostertz et al., 2004) as well as the putative MerR-type regulators YyaN and YraB (Gaballa et al., 2003) in Bacillus subtilis.
Construction of a revertant GBR8
In order to demonstrate that the failure of lm.G_1497 expression by transposon Tn917 insertion was responsible for the biofilm reduction of GB8, back mutation was carried out to construct a revertant by homologous recombination. Three revertants were successfully obtained from 607 candidates with growth of isolates on BHI plates without antibiotic at 30°C, while no growth on BHI plates containing erythromycin (erm) or chloramphenicol (cm) at 41°C. One of the revertants was named GBR8, while there were two similar fragments in both GBR8 and the wild-type L. monocytogenes 4b G (data not shown).
Biofilm formation by the Tn917 insertion mutant GB8
Tn917 insertion mutant GB8 showed significantly lower ability to form biofilm than the wild-type (Tukey's test, p<0.05). The biofilm-forming ability of the revertant GBR8 was similar to that of the wild-type (Fig. 3).

Microtiter plate assay results of biofilm formation of Listeria monocytogenes 4b G (WT [wild-type]), L. monocytogenes GB8 (Tn917 insertion mutant), and L. monocytogenes GBR8 (revertant). Biofilm was measured in 96-well microtiter plates after 3 days of inoculation in brain-heart infusion (BHI) broth. The experiments were repeated three times, and error bars indicate the standard error of the mean. Tukey's test was used to calculate the significant difference (p<0.05) in the amount of biofilm between mutant and wild-type strains as indicated with an asterisk (*).
The influence of lm.G_1497 gene on cell growth, cell motility, and cell autolysis
In order to understand the mechanism of the lm.G_1497 gene on biofilm formation, the cell growth rate, cell motility, and cell death rate (cell autolysis) were examined. GB8 exhibited the same growth rate as wild-type in BHI broth at 37°C during 24 h. Both strains entered log phase in about 7 h and stationary phase in about 11 h, at the same density. In addition, both strains showed a similar motility phenotype at both 37°C and 25°C, which suggested that there was no significant difference on the flagella function between the wild-type and the mutant strain (data not shown). In the presence of 0.05% Triton X-100, both wild-type and the mutant exhibited a decrease in OD600 during the first 5 h. Interestingly, the wild-type was more sensitive to Triton X-100 than GB8. After 5 h of treatment with Triton X-100, the percentage of the remaining wild-type cells density was approximately 40%, while that of GB8 was approximately 90% (Fig. 4). The survival of the resulting mutation GB8R in the presence of the detergent was 55%, similar to that of the wild-type strain (Fig. 4).
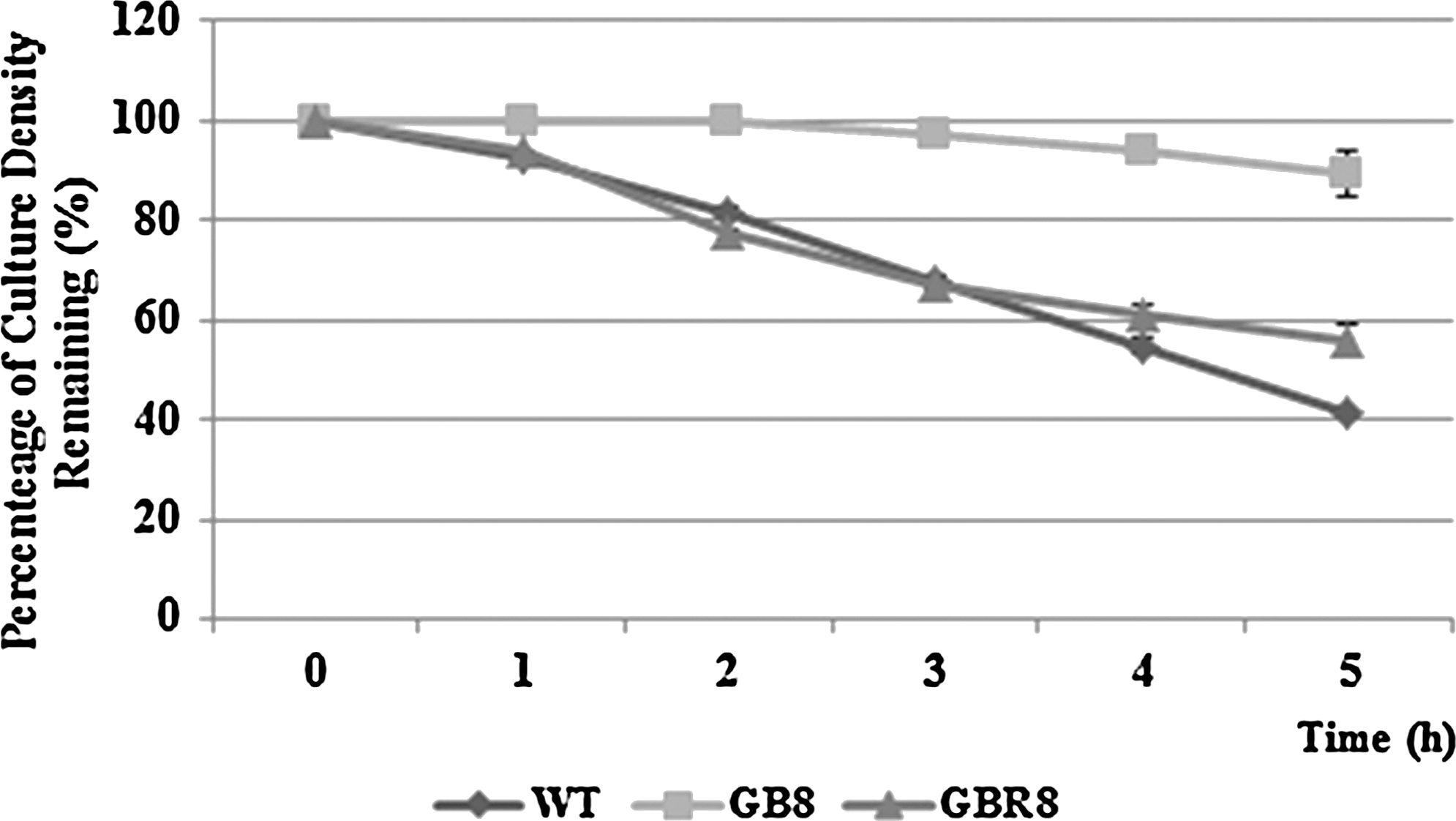
Results of Triton X-100–induced autolysis assay. Cells were mixed with 0.05% Triton X-100, and changes in OD600 were measured at various times (x-axis). The y-axis represents the percentage of culture density remaining.
Discussion
L. monocytogenes is a harmful foodborne pathogen that is capable of adhering and forming biofilm on a number of surfaces (Chae and Schraft, 2000). In this study, a gene encoding a MerR family transcriptional regulator has been identified to positively control biofilm formation in L. monocytogenes. Once the gene expression was inactivated by insertion of transposon Tn917 into L. monocytogenes 4b G chromosome, the mutant GB8 exhibited a decreased biofilm biomass.
The transposon insertion region on mutant GB8 was located 15 bp before the start codon of lm.G_1497, a MerR family regulator encoding gene. It has been reported that the promoter of MerR family transcriptional regulators is typically located 35 to 10 bp upstream of the transcriptional start site (Brown et al., 2003). In addition, quantitative real-time PCR results showed that expression of gene lm.G_1496, which is upstream of and divergently transcribed from lm.G_1497, was similar in strain GB8 and the wild-type strain. Therefore, only the expression of lm.G_1497 was inactivated by Tn917 insertion in mutant GB8.
It is known that some physiologically related genes are regulated by the MerR family members (Brown et al., 2003; Kidd et al., 2005) and numerous factors can activate the MerR family regulators (such as metal ions, oxidative stress, aldehydes, and light) and some regulators of multi-drug efflux pumps (Brown et al., 2003). Although the regulation network of the regulator encoded by lm.G_1497 is not clear, its homologous proteins SoxR (Mostertz et al., 2004) and YraB (Gaballa et al., 2003) have provided some information. SoxR is one of the well-known MerR family members, isolated from Escherichia coli, and is thought to be responsible for part of the oxidative stress response by activating transcription of the various regulons (Hancock and Klemm, 2007; Nunoshiba et al., 1992). It has also been reported that YraB in Bacillus subtilis regulates the expression of a gene related to nitric oxide (NO) stress or aldehydes sensors (Kidd et al., 2005; Nguyen et al., 2009). Since the structure of the regulator encoded by lm.G_1497 is homologous to that of SoxR and YraB, these proteins might have analogous functions. On the basis of the resistant mechanism of SoxR and YraB, it was hypothesized that an intracellular redox signal might trigger the MerR family regulator encoded by lm.G_1497 in L. monocytogenes 4b G at first, and other regulons, redox-sensors, or genes related with oxidative stress were activated by the regulator to help cell survival in stress conditions.
Biofilm formation on days 1 and 2, which only produced a small amount of biofilm (OD590<0.06), exhibited tiny differences between the wild-type and the mutant strain (data not shown), while obvious biofilm difference was observed on day 3. On the basis of the similar growth rate and death rate in wild-type and mutant strains (data not shown), the reduction of biofilm formation in the mutant strain was assumed to be unrelated with the increased death rate or reduced survival. Inside a biofilm, oxidative stress is a common condition caused by cell metabolism and environmental superoxide-generating agents, especially during the later stages of biofilm formation. The decreasing biofilm formation of mutant GB8 might result from the failure responses of some stress-related genes or sensors, which should have been activated by the regulator encoded by lm.G_1497. Besides oxidative stress condition, the long-term survival of biofilm cells could also cause limited nutrition in growing conditions (Danese et al., 2001). To survive in the adverse condition, cells would release extracellular DNA (eDNA) as an important nutrition resource in biofilm cultures by cell autolysis (Thomas et al., 2008). In order to investigate if the biofilm reduction in GB8 was caused by higher cell autolysis rate, cell autolysis induced by Triton X-100 was examined in this study. In addition, some primary factors involved in biofilm formation were examined including cell growth rate and cell motility. It was found that the mutant and the wild-type exhibited similar growth rate and motility in this study, which suggested that growth and flagella development of GB8 was not impaired. Interestingly, the autolysis rate induced by Triton X-100 in GB8 was lower than that of the wild-type (Fig. 4). The autolysis assay results may suggest that the wild-type strain sacrificed more cells to form a better biofilm than GB8 did during the later stages of biofilm formation.
In conclusion, inactivation of lm.G_1497, encoding a putative MerR family regulator conserved in Listeria species, increased biofilm formation and decreased autolysis rate induced by Triton X-100 in L. monocytogenes GB8, compared to the wild-type L. monocytogenes 4b G strain. This may suggest that this putative MerR family regulator influences cell autolysis to promote biofilm formation in L. monocytogenes. Further investigation is required to illustrate how the MerR family regulator precisely influences biofilm formation in L. monocytogenes. The role of lm.G_1497 in stress response particularly should be characterized further.
Footnotes
Acknowledgments
This work was jointly supported by the National Natural Science Foundation of China (grants 31171690, 30972485, and U1031003), the Ministry of Science and Technology of China (grants 2011DFA31220 and SS2012AA101601), and the General Administration of Quality Supervision, Inspection and Quarantine the People's Republic of China (grant 20111K147).
Disclosure Statement
No competing financial interests exist.