
Abstract
Human Noroviruses (HuNoVs) are the most frequent cause of outbreaks of acute gastroenteritis following the ingestion of raw or improperly cooked oysters. Although highly sensitive methods to detect HuNoV in oysters using reverse transcriptase–polymerase chain reaction (RT-PCR) are available, rapid methods to process samples for RT-PCR are still needed. The conventional approach is to concentrate the virus first before RNA purification to maximize assay sensitivity, but the procedures used are cumbersome. We developed a new hybridization method that is much faster and more effective compared to existing technology. The procedure includes an initial extraction of total RNA from the digestive diverticula of oysters using TRI Reagent, followed by HuNoV RNA purification using a capture probe and then HuNoV detection by real-time RT-PCR. The detection limit is approximately 100 PCR detection units of HuNoV per sample. Compared to published methods that require an initial virus concentration step before RNA extraction, the new method is much faster to complete. Approximately 3 h are needed to purify HuNoV RNA using the new method compared to at least 8 h using conventional methods. Coupled with real-time RT-PCR, the new method can detect HuNoV in contaminated oysters within 8 h. The effectiveness of the method was demonstrated using live artificially contaminated oysters and wild oysters.
Introduction
N
Although reverse transcriptase–polymerase chain reaction (RT-PCR) primers for NoV detection and quantification as well as genotyping have been available for some time now (e.g., Kojima et al., 2002; Kageyama et al., 2003; Vinje et al., 2004), a fast and efficient method to process shellfish samples for RT-PCR remains elusive. Most of the NoV RNA extraction protocols (e.g., Mullendore et al., 2001; Myrmel et al., 2004; Jothikumar et al., 2005; Kingsley, 2007; Gentry et al., 2009; Le Guyader et al., 2009) are time consuming and cumbersome, and thus deter routine testing. For example, the GPTT viral RNA extraction protocol (Kingsley and Richards, 2001) utilizes separate steps for virus elution, virus precipitation, total RNA isolation, and viral RNA purification. In a study comparing methods, Schultz et al. (2007) found that each of the three published methods included in the study (Le Guyader et al., 2000; Mullendore et al., 2001; Beuret et al., 2003) required at least 1 day to process six samples from tissue homogenization to viral RNA isolation.
In the present study, we describe a faster viral RNA extraction method that uses a biotinylated capture sequence to isolate NoV RNA from total RNA. The method eliminates the need to isolate the virus before RNA extraction and thus greatly reduces the amount of time needed to detect HuNoV in oysters using real-time RT-PCR. The new method was developed and optimized using oyster homogenates containing HuNoV GII-positive stool extract and then tested using live artificially contaminated oysters and wild oysters.
Materials and Methods
Virus stock and oyster samples
The HuNoV GII.4-positive sample was obtained from a U.S. Food and Drug Administration research laboratory. This HuNoV reference sample was added to oyster homogenates during method development and to artificially contaminate live oysters. The titer of the sample was determined by real-time RT-PCR to be 105 RT-PCR detection units (PDU)/mL, where 1 PDU is the least amount of virus required to produce a positive RT-PCR signal (also known as PCR amplifiable units, e.g., McLeod et al., 2009).
The oyster Crassostrea virginica was used throughout the study. Oyster digestive diverticula were dissected from 20 individual oysters purchased from a seafood market (Crystal Seas Seafood, Pass Christian, MS), minced with a scalpel, thoroughly mixed using a manual Potter-Elvehjem tissue grinder, and stored at −80°C until use. Digestive diverticula was used because it is where NoV is predominantly localized in oysters (Le Guyader et al., 2006; McLeod et al., 2009) and where specific binding of both GI and GII norovirus-like particles has also been demonstrated (Tian et al., 2006). To determine the detection limit of the HuNoV assay method, the stock HuNoV sample was serially diluted in PBS and then added to 100-μL subsamples of the homogenate. Each sample was prepared in duplicates and the experiment was repeated three times. Digestive diverticula homogenates without added HuNoV served as negative controls.
To compare the efficacy of the assay method to other published methods, samples of oyster homogenate containing the same amount of HuNoV were prepared and assayed. One gram of thawed oyster digestive diverticula homogenate was thoroughly mixed with 1000 PDU HuNoV, after which six 100-μL subsamples were transferred to new tubes. Two subsamples were processed using each of three RNA extraction methods. This comparison was performed three times.
To verify that the method described works, live oysters collected from a salt marsh at East Beach, Ocean Springs, MS were artificially exposed to HuNoV in the laboratory and tested. To expose oysters, the HuNoV stock diluted in PBS was first added into tanks holding 10 L of natural estuarine water (24 ppt) to reach final virus concentrations of 2000 PDU/L. Ten oysters were then added to the tank after 1 h, during which water was continuously circulated using a small submersible pump. Oysters in a tank without added HuNoV served as negative control. Three oysters were sampled after 1 and 3 days. As was done throughout the study, only the digestive diverticula were assayed. After dissection, 100 μL of the minced tissue (representing approximately 10% of the entire sample) were tested for the presence of HuNoV as described below.
Oysters tested to determine whether HuNoV could be detected in field populations using the present method were collected from a marina in Ocean Springs, and salt marshes in Ocean Springs and Pass Christian, MS.
NoV RNA isolation
Total RNA was first extracted from oyster digestive diverticula using the single-step phenol-guanidinium thiocyanate extraction method with TRI Reagent (Chomczynski, 1993) according to the manufacturer's protocol. To isolate HuNoV RNA, the RNA pellet was dissolved in 60 μL of diethyl pyrocarbonate (DEPC)-treated water, heated at 94°C for 5 min, and then quick chilled on ice for 5 min. Afterwards, 2 μL of 1 μM biotinylated COG2R probe (Table 1), 1 μL of RNasin Plus RNase Inhibitor (Promega Corp, Madison, WI), and 7 μL of 10X hybridization buffer (0.4 M NaCl, 40 mM PEPES, 1 mM EDTA, pH 6.6) were added to each 60-μL RNA sample. To facilitate hybridization of HuNoV RNA to the biotinylated COG2R probe, samples were incubated at 45°C for 1 h with continuous agitation. To determine the optimum amount of time to use, times ranging from 1 min to 5 h were tested.
Nucleotide positions based on Lordsdale virus (Genbank accession no. X86557).
BIO, biotin; FAM, fluorescein; BHQ, Black Hole Quencher.
To capture HuNoV RNA hybridized to the biotinylated probe, 10 μL of washed streptavidin-coated magnetic beads (Dynabeads MyOne™ Streptavidin C1, Life Technologies, Grand Island, NY) and 5.9 μL of DEPC-treated 5 M NaCl were added and the samples were mixed continuously for 30 min at 25°C. Times ranging from 1 min to 2 h were tested to determine the amount of time to use for maximum probe recovery. To remove contaminants, the magnetic beads were concentrated to one side of the tube using a magnet and washed three times with 100 μL of 1X washing buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, and 1 M NaCl). To elute the captured HuNoV RNA, 10 μL of DEPC-treated water was added and the sample was heated at 94°C for 5 min. After brief centrifugation, the magnetic beads were captured again using the magnet, and the supernatant containing HuNoV RNA was transferred to a new tube for analysis by real-time RT-PCR. DEPC-treated water and RNA extracted directly from the HuNoV reference sample were used as negative and positive controls, respectively. The efficacy of HuNoV detection using the present RNA isolation method was compared to those achieved using two published methods with slight modifications. With the method of Beuret et al. (2003), TRI Reagent was used to extract RNA instead of QIAamp Viral RNA Mini Kit (Qiagen Inc., Valencia, CA). With the method of Baert et al. (2007), QIAamp Viral RNA Mini Kit was used in place of the RNeasy Mini Kit (Qiagen Inc.). HuNoV was added to oyster digestive diverticula homogenates, mixed, and then aliquoted so that each subsample contained 100 PDU. Two subsamples were extracted using each of the three methods, and the experiment was performed three times. The amount of HuNoV RNA isolated using the three methods was compared by real-time RT-PCR.
Detection of HuNoV using real-time RT-PCR
Primers used for reverse transcription and real-time PCR of HuNoV cDNA are listed in Table 1. cDNA was synthesized using 5.75 μL of purified RNA, 1 μM primer, 0.5 mM of each dNTP, 10 U of RNasin Plus RNase Inhibitor (Life Technologies), 1.5 mM of MgCl2, 0.5 μL ImProm-II Reverse Transcriptase (Promega Corp., Madison, WI) in a total volume of 10 μL. RNA denaturation took place at 70°C for 5 min, primer annealing at 25°C for 5 min, DNA extension at 42°C for 60 min, and reverse transcriptase inactivation at 70°C for 15 min. Water was included as a negative control for both reverse transcription and PCR. Serially diluted plasmid DNA containing cloned HuNoV cDNA (described below) was used as standards during real-time PCR. Amplification reactions contained 2 μL cDNA, 400 nM each primer (JJV2F and COG2R, Table 1), 200-nM probe (RING2-TP), 0.5 U DNA polymerase, and 200 μM dNTPs in a total volume of 10 μL. cDNA amplification was carried out using a melting step at 95°C for 10 min followed by 45 cycles of 95°C for 15 s to melt cDNA and 60°C for 60 s to anneal and extend primers. The size of the amplicon was 98 base pairs.
The plasmid DNA serially diluted and used as standards during real-time PCR experiments was obtained by cloning the RT-PCR product amplified using the HuNoV reference RNA described above. RNA was reverse transcribed using random hexameric primers and AMV Reverse Transcriptase and then amplified using the primers JJV2F and COG2R (Table 1). Cloning was performed using the vector pSMART HCKan and host cell Escherichia cloni 10G (Lucigen Corp., Middleton, WI) as instructed by the manufacturer. Multiple aliquots of purified plasmid DNA from a single preparation were stored at −80°C. An aliquot was thawed for each experiment and serially diluted 10-fold to obtain standards containing 102–107 genome copies of HuNoV cDNA.
Statistical analysis
The statistical significance (p<0.05) of differences in the amount of HuNoV detected using the three RNA isolation methods and the different hybridization conditions were evaluated using one-way analysis of variance (SPSS version 13.0). Tukey's HSD test was used to test the significance of differences among means.
Results
Method development
Real-time RT-PCR results from initial experiments showed that the use of longer probe-target hybridization times, up to 5 h, and probe capture times, up to 2 h, resulted in slightly reduced assay sensitivity as indicated by the higher threshold cycle (Ct) (data not shown). Further experimentation showed that additional savings in time could be achieved by shortening the hybridization and capture times. The assay sensitivity was not significantly different among hybridization times of 1, 15, and 60 min and probe captures times of 10 and 30 min (Fig. 1). However, reducing the probe capture time to 1 min reduced the assay sensitivity significantly (p<0.05), resulting in Ct values that increased by approximately four cycles (Fig. 1).
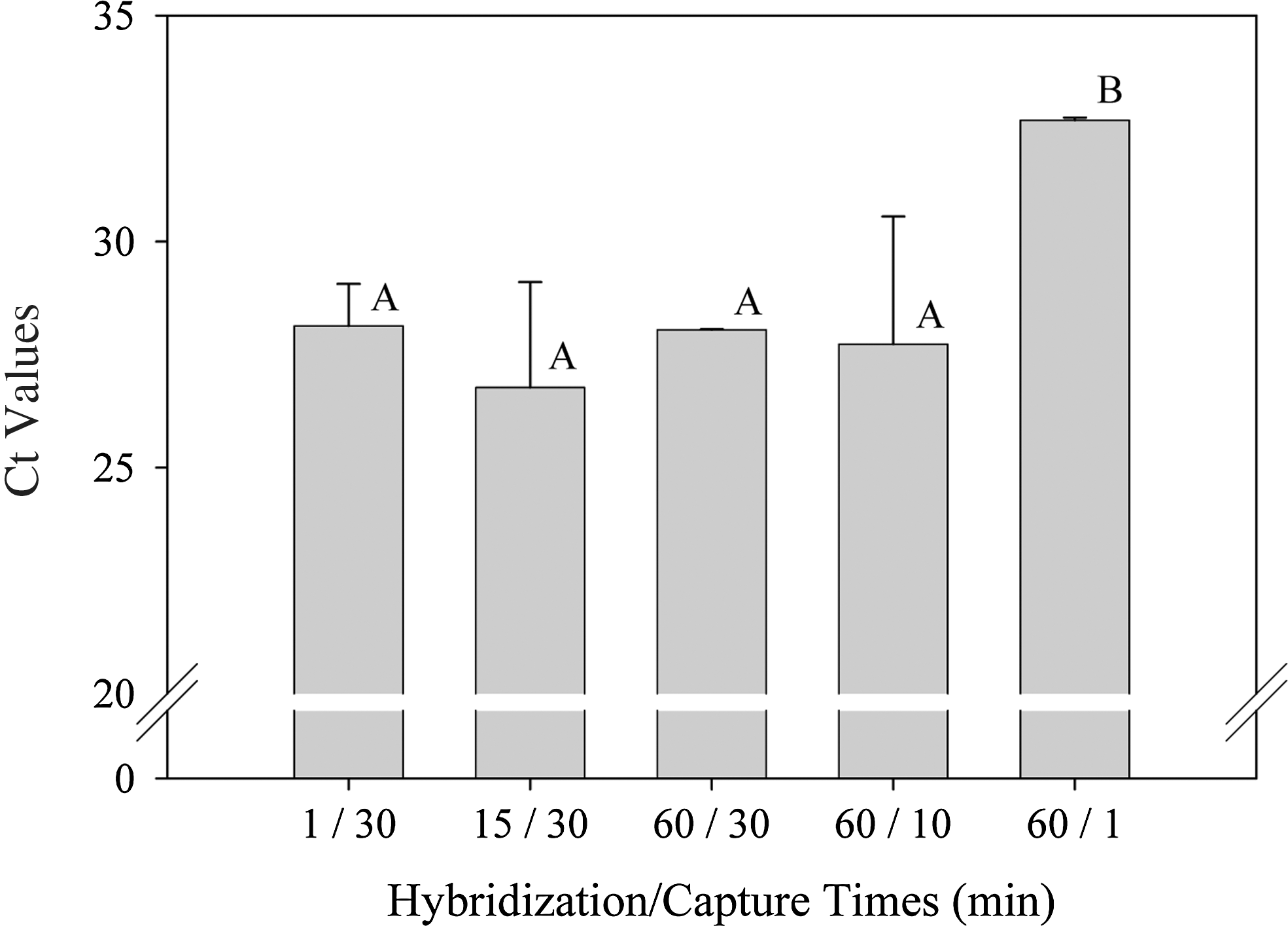
The effect of different probe hybridization and capture times on threshold cycle (Ct) values during human Norovirus detection by real-time reverse transcriptase polymerase chain reaction. Mean Ct values with the same letter are not statistically different. Error bars indicate standard deviation.
Detection of HuNoV in oysters
The lower detection limit of the method was approximately 10 PDU. HuNoV was not detected in samples containing 1 PDU but was detected in 9 of 12 samples containing 10 PDU and in 12 of 12 samples containing 100 PDU HuNoV (Table 2). Among live oysters artificially contaminated with HuNoV, we were able to detect the virus in all three of the oysters exposed for 1 day and in two of the three oysters exposed for 3 days. Oysters not exposed to HuNoV were all RT-PCR negative. Among wild oysters, 3 of 44 collected from Davis Bayou, Ocean Springs, MS were found to be contaminated with HuNoV (Table 3). Two HuNoV-contaminated oysters were collected on July 13, 2009 and one on October 28, 2009. Oysters collected from Pass Christian and the Ocean Springs Harbor were RT-PCR negative for HuNoV.
PDU, transcriptase–polymerase chain reaction detection units.
Oyster homogenates contained different amounts of HuNoV indicated.
Comparison to two other RNA extraction methods
In terms of processing time, it took approximately 3 h to prepare RNA for RT-PCR with the probe-hybridization method using probe hybridization and bead capture times of 1 h and 30 min, respectively. In contrast, only 1.5 h was needed using the total RNA approach of Baert et al. (2007) and at least 8 h using the approach of Beuret et al. (2003), whereby virus particles were isolated first before RNA extraction. In terms of HuNoV detection, the mean Ct value obtained was 25.6±0.4 using RNA extracted with the target-capture method and 31.4±0.2 using RNA isolated with the Beuret et al. (2003) method. We were not able to detect HuNoV in the total RNA extracted using the method of Baert et al. (2007).
Discussion
Consumption of shellfish contaminated with NoV has long been recognized as a problem worldwide. To better monitor contamination, highly sensitive molecular assays to detect HuNoV are available (e.g., Kageyama et al., 2003; Jothikumar et al., 2005; Kou et al., 2006). However, an effective method to first isolate NoV RNA for subsequent detection by one of the molecular assays is still urgently needed. A frequently used approach is to isolate the virus first followed by RNA isolation and NoV detection by RT-PCR (e.g., Jaykus et al., 1996; Beuret et al., 2003; Comelli et al., 2008; Le Guyader et al., 2009; DePaola et al., 2010). This approach allows the use of large tissue masses, sometimes multiple oysters in one homogenized sample, thereby enhancing the probability of detecting NoV when prevalence of contaminated oysters or when virus concentration in each oyster is low. However, the multiple steps and large amount of time required to process samples makes the approach impractical for routine monitoring.
To speed up sample processing, some use total RNA extracted from shellfish samples for NoV detection by RT-PCR (e.g., Baert et al., 2007; Boxman et al., 2006). Using this faster approach, assay sensitivity is sacrificed because the amount of RNA used in the initial reverse transcript step for RT-PCR is limited and most of the RNA is likely to be oyster RNA. This may explain why we were not able to detect HuNoV in total RNA extracted from oyster homogenates containing 100 PDU HuNoV using the approach of Baert et al. (2007).
To increase sensitivity while retaining the faster speed of the direct RNA extraction approach, we incorporated a probe hybridization technique to capture and isolate NoV RNA before RT-PCR. The probe sequence, adapted from Kageyama et al. (2003), was selected to capture diverse GII NoV genotypes as it resides at the ORF1-ORF2 junction, the most conserved region in the Norwalk-like genome (Katayama et al., 2002). The sequence has been shown by Kageyama et al. (2003) to be present in over 30 different strains of GII noroviruses from divergent sources. The method we describe is made faster by eliminating the initial virus isolation step. Total RNA is first purified from oyster tissue and then HuNoV RNA is selectively captured using a biotinylated DNA probe that hybridizes to HuNoV RNA. The biotinylated DNA:RNA duplex is isolated using streptavidin coated magnetic beads to immobilize the duplex while oyster RNA is removed. The HuNoV RNA is released by heating the DNA:RNA bead complex and then detected by real-time RT-PCR. The entire procedure can be completed in 3 h using probe hybridization and bead capture times of 1 h and 30 min, respectively. We discovered during later experiments that there was no difference in assay sensitivity between probe hybridization times of 1 and 60 min and bead capture times of 10 and 30 min. This suggests that it may be possible in the future to process samples much more quickly than 3 h by hybridizing for 1 instead of 60 min and to capture the beads for 10 instead of 30 min.
We showed that the assay procedure works using oysters artificially contaminated in the laboratory and wild oysters. The detection of HuNoV in three wild oysters was surprising because routine monitoring of enterococci as an indicator of fecal pollution at a sampling site (Station 15) 1 km away by the Mississippi Department of Environmental Quality rarely indicated a problem. Online data show that among the weekly samples in 2009, only five exceeded the threshold for a beach advisory (104 CFU/dL) during the entire year (MSDEQ, 2009). This apparent lack of correlation between fecal indicators and norovirus presence in environmental waters has been reported by others (Hernandez-Morga et al., 2009; Yang et al., 2012).
An extraction and/or amplification control is often included for food samples to assess the possible presence of PCR inhibitors (LeGuyader et al., 2009; Diez-Valcarce et al., 2011). One was not included in the present study when whole oysters were used to test the efficacy of the assay procedure but should be included in future routine monitoring efforts. It was noticeable, however, that initial total RNA extracted from oyster diverticula were often pigmented whereas final extracts after probe capture and release were always clear and colorless.
During method development, we noticed that the source of polypropylene microcentrifuge tube used makes a profound difference in the sensitivity of the assay. Those that work well can be easily distinguished. After the streptavidin magnetic beads have been added to capture the biotinylated DNA:RNA duplex, the uniformly brown solution should become clear quickly when the tube is placed against a magnet. With tubes that work well, the vast majority of beads congregate on the side of the tube next to the magnet. With tubes that do not work well, often the more expensive “low adhesion” tubes, a noticeable amount of the brown magnetic beads adhere elsewhere inside the tube. We surmise that some type of attractive force between either biotin or streptavidin and plastic treated to minimize adhesion of nucleic acids prevents complete recovery of captured HuNoV RNA.
Conclusion
The probe hybridization procedure described in this study provides a fast method to isolate HuNoV RNA from oyster tissues for detection by RT-PCR. The protocol differs from other methods by using a biotinylated-DNA capture probe to isolate a specific viral RNA target from total RNA instead of isolating the virus itself before RNA extraction. This approach can be adopted for the simultaneous isolation and detection of other RNA viruses of concern in shellfish such as rotavirus and hepatitis A virus by incorporating additional target-specific capture probes.
Footnotes
Acknowledgments
The HuNoV GII-positive reference sample was kindly provided by Dr. Jacquelina Woods, U.S. Food and Drug Administration, Dauphin Island, AL. This study was funded by grants from the Mississippi Coastal Impact Assistance Program, Project M09AF16192 and the U.S. Environmental Protection Agency, Gulf of Mexico Program, Project MX96429505-0.
Disclosure Statement
No competing financial interests exist.