
Abstract
Cronobacter spp. (formerly Enterobacter sakazakii), a foodborne pathogen linked to powdered infant formula, is a rare cause of invasive infection with a high mortality rate in neonates. In this study, the Cronobacter sakazakii ATCC 29544 and C. muytjensii ATCC 51329 glutaredoxin 2 (grxB) genes were cloned and sequenced. Based on the unique regions of the Cronobacter grxB genes, two primers were synthesized to develop and optimize a Cronobacter-specific polymerase chain reaction (PCR) method. The PCR assay amplified a 378-bp DNA product from all positive controls, which are composed of 45 strains of Cronobacter spp., but not from any of 45 non-Cronobacter bacterial strains. The detection limits of this method are 104 colony-forming units (CFU)/mL of Cronobacter spp. in infant formula directly and 100 CFU/mL after an 8-h enrichment step. In summary, we have developed a PCR assay based on the grxB sequence. Combined with enrichment culturing, this technique offers a rapid and sensitive method for the detection of Cronobacter spp.
Introduction
T
For the detection of Cronobacter in PIF, the International Standards Organization (ISO) has published a standardized method that is a considerable improvement on the original method recommended by the U.S. Food and Drug Administration. However, the method is time consuming, labor intensive, and sometimes hampered by false-negative or false-positive results due to its reliance on pigment production and the biochemical profile of isolates (Guillaume-Gentil et al., 2005). A number of conventional polymerase chain reaction (PCR) and real-time PCR systems for the rapid detection and identification of Cronobacter species based on different target regions, including the 16S rRNA gene (Keyser et al., 2003; Lehner et al., 2004; Hassan et al., 2007), 16S-23S rDNA internal transcribed spacer (Liu et al., 2006), α-glucosidase genes (Lehner et al., 2006a; Lehner et al., 2006b), outer membrane protein A gene (Mohan Nair and Venkitanarayanan, 2006), zinc-containing metalloprotease gene (zpx) (Kothary et al., 2007), and macromolecular synthesis operon (Seo and Brackett, 2005), have been developed and are intended to provide rapid detection and identification of Cronobacter. Glutaredoxin 2 (grxB) in Escherichia coli catalyzes glutathione–disulfide oxidoreductions (Vlamis-Gardikas et al., 1997). The grxB gene sequence was found to be specific for Cronobacter after comparison of the published nucleotide sequences of grxB from Cronobacter with other bacteria. In this study, we report the identification and molecular cloning of the grxB gene in Cronobacter spp. and the development of a PCR technique based on the grxB gene sequence for the specific identification of Cronobacter spp. in pure culture and in reconstituted infant formula.
Materials and Methods
Bacterial strains and DNA extraction
Strains used in the present study are listed in Table 1. All of the Cronobacter strains were isolated from PIF or other food, following the ISO/TS 22964:2006 method and identified using the API 20E biochemical system (API 20E; bioMerieux, Marcy l'Etoile, France). The bacterial genomic DNA samples were extracted using the SK1201 DNA extraction kit (Sangon Biotech, Shanghai, China) and then stored at −20°C for use.
PCR reaction and electrophoresis conditions
Fifty-microliter PCR amplification reaction mixtures contained 25 μL of 2×Taq PCR Master Mix (LifeFeng, Guangzhou, China), 2 μL each of 10-μM primer, 2 μL of template DNA (∼70 ng), and sterile, deionized water. The PCR was performed using a T-Professional thermocycler (Biometra, Goettingen, Germany) under the following conditions: a 5-min denaturation at 95°C, followed by 40 cycles of 95°C for 30 s, 58°C for 45 s, and 72°C for 60 s, with a final amplification at 72°C for 5 min. A 6-μL aliquot of each amplicon was resolved on a 1% agarose gel with GoldView™ Nucleic Acid Stain (SBS Genetech Co., Ltd., Beijing, China); the gels were examined using the ImageQuant 350 system (GE Healthcare, Chalfont St. Giles, UK).
Cloning and sequencing of the grxB gene from Cronobacter spp.
The PCR primers CSEGBF and CSEGBR (Table 2) were designed according to the grxB gene sequence in the completed C. sakazakii ES15 genome (GenBank accession no. FN543093) and synthesized by BGI (Shenzhen, China). DNA fragments of 663 bp were amplified from C. sakazakii ATCC 29544 and C. muytjensii ATCC 51329. The PCR reaction conditions were the same as those described above.
Italics refer to the Vibrio parahaemolyticus segment.
The amplified product (50 μL) was characterized by electrophoresis on a 1% agarose gel and then purified with a TaKaRa MiniBEST Agarose Gel DNA Extraction Kit (TaKaRa Biotech). The pure amplicons were cloned into the pMD18-T vector (TaKaRa Biotech) and then transformed into TOP10 cells; transformed TOP10 cells carrying the recombinant plasmid were selected by blue–white screening and ampicillin resistance. The cloned products were sequenced by BGI (Shenzhen, China) using primers for pMD18-T (M13F and M13R). The sequences, based on bidirectional sequencing of the same cloned product, were compiled using Vector NTI Suite8 (InforMax, Bethesda, MD). The compiled sequences were analyzed by MegAlignin DNAStar software (DNASTAR Inc., Madison, WI), in comparison to related sequences downloaded from the National Center for Biotechnology Information.
Primer design for Cronobacter-specific PCR
Based on the data for seven sequences (Fig. 1) and the results of the multiple alignment analysis, two sets of primers (CGB and CGB1) for the grxB gene were designed using conserved region of Cronobacter spp.; these primers were evaluated for species specificity using the BLAST program on the National Center for Biotechnology Information homepage. Two sets of primers were synthesized (BGI, Shenzhen, China) to amplify 378-bp (CGBF and CGBR) and 301-bp (CGB1F and CGB1R) fragments of the grxB gene.

Alignment of the nucleotide sequences of Cronobacter sakazakii ATCC 29544 and C. muytjensii ATCC 51329 grxB with the corresponding gene sequences from other bacteria. 29544, C. sakazakii ATCC 29544; ES15, C. sakazakii ES15; z3032, C. turicensis z3032; 51329, C. muytjensii ATCC 51329; MG1655, Escherichia coli K-12 MG1655; Sd197, Shigella dysenteriae Sd197; 9150, Salmonella enterica subsp. enterica serovar Typhimurium Ty2. The boxed sequences indicate the region bound by the specific primers CGBF and CGBR.
After screening the specific primers in Cronobacter spp., the internal amplification control (IAC) was designed based on the specific primers. The 50-μL PCR reaction mixtures for the Cronobacter-specific test contained 2 μL each 10 μM CGBF and CGBR primer (Table 2), 25 μL 2×Taq PCR Master Mix (LifeFeng, Guangzhou, China), 2 μL (∼70 ng) genomic DNA, and 0.07 fg of IAC plasmid (corresponding to approximately 100 copies). The thermal cycling and electrophoresis conditions were the same as those described above.
Internal amplification control for Cronobacter-specific PCR
An IAC was designed as a positive control for every reaction mixture to ensure that a negative result was due to the absence of target sequences rather than to an inhibition of the reaction (Hoorfar et al., 2004). The IAC target DNA consisted of a 509-bp fragment of the Vibrio parahaemolyticus thermolabile hemolysin (tlh) gene flanked by the target sequences of the CGBF and CGBR primers (Table 2). The amplification reaction mixtures (50 μL) contained 1 μL each of 10 μM IAC-VPF and IAC-VPR primer (Table 2), 25 μL of 2×Taq PCR Master Mix (LifeFeng, Guangzhou, China), and 1 μL (∼35 ng) of V. parahaemolyticus ATCC 33847 genomic DNA. The thermal cycling conditions were the same as those described above.
The product (50 μL) amplified in the IAC reaction was cloned (see section Cloning and Sequencing of the grxB Gene from Cronobacter spp.). The IAC plasmid was extracted from TOP10 cells using the Plasmid Miniprep Kit (Biomiga, San Diego, CA). The concentration of the IAC plasmid was determined using a Bio Spec-nano spectrophotometer (Shimadzu, Kyoto, Japan), with serial dilutions performed using 10 mM Tris-HCl (pH 8.5). The concentration of the IAC plasmid to be added to the PCR mixtures was optimized from the different template preparations used in the individual experiments to allow the efficient amplification of the Cronobacter-specific PCR product.
Detection limit of Cronobacter spp. in pure culture
One colony of C. sakazakii ATCC 29544 on a tryptic soy agar plate was selected and cultured in 10 mL tryptic soy broth for 12 h at 37°C; serial 10-fold dilutions (108–100 CFU/mL) were performed using sterile deionized water. Aliquots of each dilution (1 mL) were transferred to 1.5-mL microcentrifuge tubes and centrifuged at 16,000×g for 10 min, and 800 μL of the supernatant was removed. The cell pellets were resuspended in 200 μL of sterile deionized water, placed in a 100°C water bath for 10 min, and centrifuged in a tabletop microcentrifuge (Sigma 3K30, Sigma–Aldrich Corp., St. Louis, MO) at 6000×g for 3 min. A 10-μL aliquot of the supernatant of each dilution was used as the template in a reaction mixture (50 μL) containing 25 μL 2×Taq PCR Master Mix (LifeFeng), 0.8 μL each of 10-μM primer (CGBF and CGBR), and 0.07 fg (corresponding to approximately 100 copies) IAC.
Detection limit of Cronobacter spp. in artificially contaminated PIF
A test of artificially contaminated PIF was conducted to verify the feasibility of the PCR. Three different brands of PIF were obtained from local supermarkets and were confirmed to be negative for Cronobacter spp. by the ISO/TS 22964:2006 method. The PIFs were reconstituted by mixing 1-g aliquots in 8 mL sterile distilled water in 15-mL sterile tubes. Serial 10-fold dilutions of a C. sakazakii ATCC 29544 cultures were generated using sterile distilled water, and 1-mL volumes were added to 9 mL reconstituted PIF to obtain final concentrations of C. sakazakii ranging from 108 to 100 CFU/mL. DNA was extracted from PIF spike-in samples either directly or after an 8-h enrichment step at 37°C. Aliquots of the PIF spike-in samples (1 mL) were centrifuged at 16,000×g for 10 min, and the supernatant was removed; any supernatant remaining in the tube was removed using a sterile cotton swab. The pellets were resuspended in 200 μL sterile distilled water, and the samples were placed in a 100°C water bath for 10 min and centrifuged at 6000×g for 3 min. A 10-μL aliquot of the supernatant was used as the template in a 40-cycle PCR under the above-described PCR and electrophoresis conditions.
Detection limit of Cronobacter spp. in the presence of Salmonella Typhimurium
Salmonella has also been reported to be an intrinsic contaminant of PIF. To investigate whether the presence of both Salmonella and Cronobacter would affect the sensitivity of the PCR, we used DNA from artificially contaminated PIF samples containing different levels of C. sakazakii ATCC 29544 and Salmonella Typhimurium CMCC 50115 as the template for PCR. In these experiments, 10-mL aliquots of reconstituted PIF samples containing C. sakazakii ATCC 29544 (108–100 CFU/mL) were inoculated with Salmonella Typhimurium CMCC 50115 (107 CFU/mL). Then, 1-mL aliquots were collected from each sample and processed as described for the artificially contaminated PIF.
Nucleotide sequence accession numbers
The grxB nucleotide sequences of the two strains were submitted to the GenBank database under the accession numbers JX129376 (C. sakazakii ATCC 29544) and JX129377 (C. muytjensii ATCC 51329).
Results
PCR analysis and primer design for the detection of Cronobacter spp.
A comparison of the nucleotide sequences of grxB from C. sakazakii ATCC 29544 and C. muytjensii ATCC 51329 with the published sequences of C. sakazakii ATCC BAA-894 (CP000783), C. sakazakii ES15 (CP003312), C. turicensis z3032 (FN543093), E. coli K-12 MG1655 (NC_000913), Shigella dysenteriae Sd197 (NC_007606), and Salmonella enterica subsp. enterica serovar Typhimurium Ty2 (NC_004631) is shown in Figure 1.
Two primer pairs—CGBF-CGBR and CGB1F-CGB1R—were designed based on the nucleotide sequence of grxB, and the primer sequence exhibited 100% identity with the Cronobacter but was not homologous to the E. coli K-12 MG1655, Sh. dysenteriae Sd197, and Sa. enterica subsp. enterica serovar Typhimurium Ty2 sequences. The specificities of the primers were evaluated using 45 Cronobacter and 45 non-Cronobacter strains (Table 1). The CGBF-CGBR primer pair was found to be specific for Cronobacter. Primer CGBF was complementary to a region from nucleotide 20 to nucleotide 47 of the cloned grxB fragment, whereas CGBR was complementary to a region from nucleotide 373 to nucleotide 397 of the cloned grxB fragment. When amplified using the primer pair CGBF-CGBR, the 45 Cronobacter strains all yielded the expected product of 378 bp, whereas none of the other strains produced a positive signal. However, the 562-bp IAC product was amplified using template DNA from the 45 non-Cronobacter negative control strains, ruling out the possibility of false negatives due to PCR inhibition (Fig. 2).
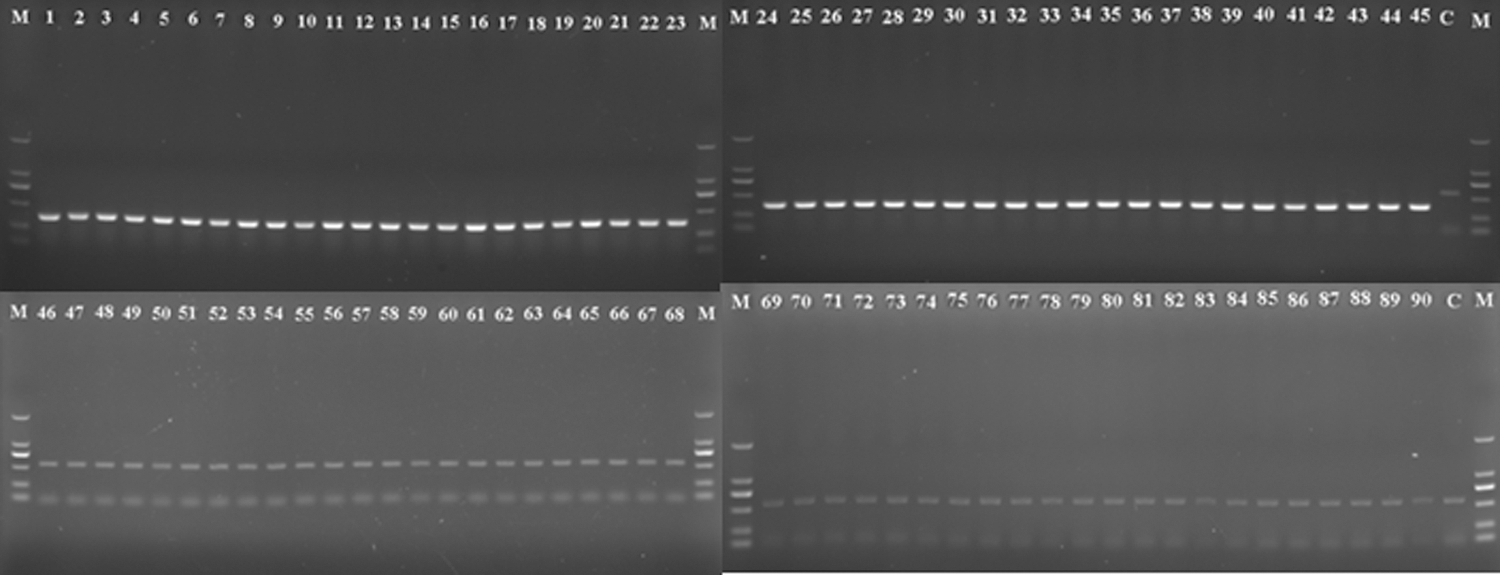
Detection of Cronobacter spp. by polymerase chain reaction (PCR) with the specific primers CGBF and CGBR. The PCR products were analyzed by electrophoresis in a 1% agarose gel. A 378-bp DNA fragment was amplified from the grxB gene of Cronobacter, and a 562-bp DNA fragment was amplified from the internal amplification control (IAC). Lane M: molecular-weight marker (DL2000 LadderMix; TaKaRa Biotech, Tokyo, Japan); lanes 1–45: PCR products from Cronobacter strains 1–45, as listed in Table 1; lanes 46–90: PCR products from negative control DNA from bacterial strains 46–90, as listed in Table 1; C, control PCR without any template DNA.
Detection limit of the PCR assay in pure culture and in artificially contaminated PIF
Tenfold serial dilutions of a pure culture (108–100 CFU/mL) were generated to determine the detection limit of the PCR technique, which was found to be 103 CFU/mL in pure culture (Fig. 3A).
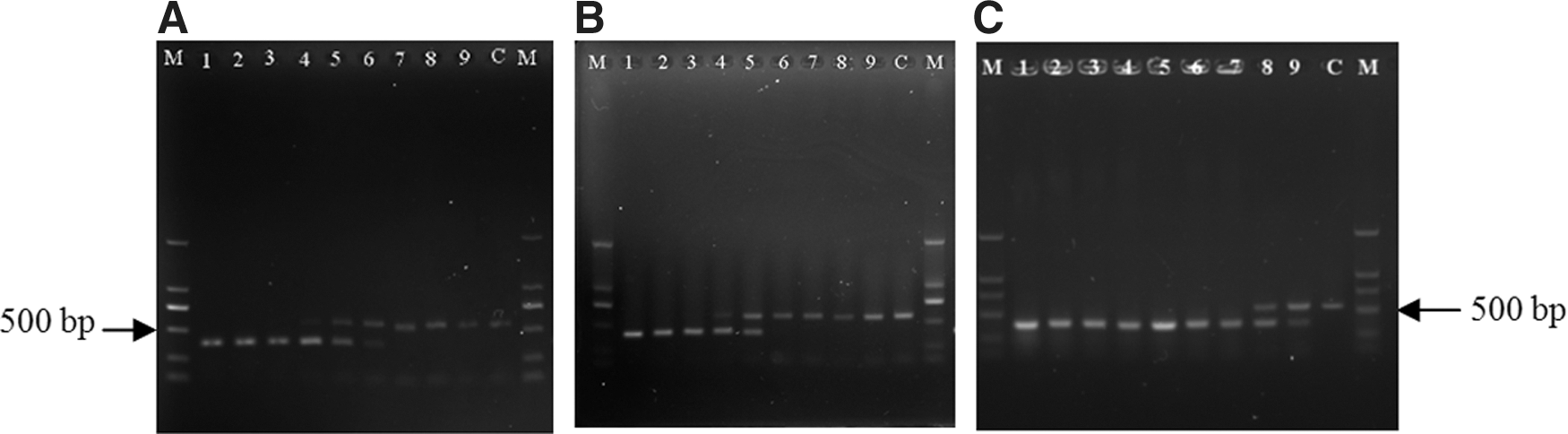
The detection limits of the polymerase chain reaction (PCR) assay.
Three different brands of PIF were used to assess the detection limit of the PCR assay in food matrices. We found that 104 CFU/mL could be detected by the direct amplification of infant formula samples spiked with Cronobacter (Fig. 3B); the incorporation of an enrichment step prior to amplification enhanced the sensitivity of the PCR. After 8 h of enrichment, the PCR assay was able to detect 100 CFU/mL of bacteria in reconstituted PIF (Fig. 3C). Only the IAC band was observed when the concentration of Cronobacter was below the detection limit of the PCR assay (Fig. 3).
Detection limit of the PCR assay in the presence of Salmonella
The presence of Salmonella did not affect the detection limit of the PCR for detecting Cronobacter in PIF. The target grxB sequence was amplified from all PIF samples containing 108–104 CFU/mL of C. sakazakii ATCC 29544 when inoculated with 107 CFU/mL of Salmonella Typhimurium CMCC 50115. However, no positive signal was observed for the samples containing 103–100 CFU/mL of C. sakazakii and negative control samples (PIF with 107 CFU/mL of Salmonella Typhimurium but no C. sakazakii).
Discussion
In this study, we cloned and sequenced the grxB genes of C. sakazakii ATCC 29544 and C. muytjensii ATCC 51329. Analyses of the genomic sequences of members of the Cronobacter genus available in GenBank, including C. sakazakii ES15 (CP003312), C. turicensis z3032 (FN543093), and C. sakazakii ATCC BAA-894 (CP000783), revealed that the grxB gene is located between mdtH, which encodes a multidrug resistance protein, and yceB, which encodes a lipoprotein. The CSEGBF and CSEGBR primers (Table 2) were designed to amplify an approximately 663-bp region encompassing the grxB gene from C. sakazakii ATCC 29544 and C. muytjensii ATCC 51329.
The specific primers for Cronobacter were designed based on sequence alignment and were analyzed using the BLAST database. No non-Cronobacter DNA sequence in the BLAST database exhibited any identity to the primers. This set of primers was also evaluated for Cronobacter specificity using DNA purified from 45 Cronobacter strains and 45 non-Cronobacter strains. The PCR amplified a 378-bp DNA fragment from all the Cronobacter strains, and no false-positive results were found among any of the non-Cronobacter strains. These results indicated that the primers and the PCR assay are specific for Cronobacter.
The use of IAC is required for the detection of foodborne bacteria by PCR in food products to prevent false-negative results that might be caused by PCR inhibition. However, the concentration of the IAC template is a critical factor (Hoorfar et al., 2004): Excess IAC template can compete with the target PCR, leading to false-negative results. Therefore, the lowest amplifiable IAC template concentration must be determined carefully and empirically. In this study, we designed a competitive IAC template with a product (562 bp) that could be amplified using the same primer set used to amplify the target product (378 bp), and we found that the lowest successfully amplified IAC template concentration was 0.07 fg/assay. Due to competition, only the species-specific grxB product was produced at this concentration of IAC when the samples contained Cronobacter genomic DNA (Fig. 2). Moreover, the IAC product was amplified in all samples containing non-Cronobacter DNA (Fig. 2), ruling out false-negative results due to amplification failure. The inclusion of IAC did not affect the detection limit of the PCR in pure culture or artificially contaminated PIF, in either the presence or absence of Salmonella Typhimurium. In these experiments, the PCR detection limit in the presence of the IAC template was similar to that in the absence of IAC. These results demonstrate that the IAC template concentration of 0.07 fg/assay is appropriate.
The detection limit was 103 CFU/mL using pure culture. However, nontarget background microflora may affect the detection limit of PCR using PIF (Mohan Nair and Venkitanarayanan, 2006), and different brands of PIF may have different indigenous microflora owing to the differences in ingredients and manufacturing protocols. Therefore, we tested three different brands of PIF from three different manufacturers in our study. The detection limit of the PCR using artificially contaminated PIF was 104 CFU/mL without enrichment, and there were no differences among the three brands. However, when the artificially contaminated samples were cultured at 37 °C for 8 h before being subjected to PCR, the detection limit increased to 100 CFU/mL. Moreover, our results also show that the detection limit of the PCR assay was not reduced even in the presence of nontarget bacteria such as Salmonella Typhimurium at levels as high as 107 CFU/mL.
Conclusions
In the present study, the glutaredoxin 2 genes (grxB) of C. sakazakii ATCC 29544 and C. muytjensii ATCC 51329 were cloned and sequenced. A PCR assay based on the grxB gene was developed for the rapid detection of Cronobacter in PIF, and this assay unequivocally distinguished Cronobacter spp. from other bacterial species. This technique can serve as a tool for rapid screening of food samples (after enrichment) or as a confirmation step when colonies are obtained from agar plates. Another advantage of this assay is that it incorporates an internal positive control, which controls for false-negative results that might be caused by PCR inhibitors. In summary, this PCR method, combined with enrichment culturing, is surely worthy of committed developmental effort and can be widely used to detect Cronobacter spp. in PIF or other food samples.
Footnotes
Acknowledgments
We acknowledge the financing support of National Natural Science Foundation of China, No.U1031003 and the Cooperation Project in Industry, Education and Research of the Ministry of Education in Guangdong Province, No. 2011B090300077.
Disclosure Statement
No competing financial interests exist.