
Abstract
Rapid and easy-to-use molecular subtyping methods are being explored and used for the surveillance of bacterial diseases, including multiple-loci variable number of tandem repeats (VNTR) analysis (MLVA). In this study, we assessed different VNTR combinations for the subtyping of Vibrio cholerae serogroups O1 and O139 with strain panels selected from a long-term nationwide cholera survey. By only using three highly variable loci (VC0147, VCA0171, and VCA0283), we acquired a high discriminatory power, which equals that found after using a combination of all nine loci and that of a pulsed-field gel electrophoresis analysis. Evaluation using the outbreak strains showed a good clustering of the three-loci MLVA (VC0147, VCA0171, and VCA0283). In addition, a six-loci MLVA (VC0147, VC0437, VC1457, VC1650, VCA0171, and VCA0283) protocol allowed for the clustering of O1/O139 V. cholerae strains, which have different serogroups/biotypes and toxigenic/nontoxigenic characteristics. Here, we propose that the three-loci MLVA can be utilized as a molecular subtyping protocol in cholera epidemiological investigations, and the six-loci MLVA can be used in phylogenetic and population structure analyses of V. cholerae O1/O139.
Introduction
C
MLVA is frequently applied for genotyping of bacterial pathogens due to its simplicity and high discriminating power (Lindstedt, 2005). The MLVA results between different laboratories may be easily compared by the exchange of the allele profiles (Onteniente et al., 2003). Danin-Poleg et al. (2007) and Stine et al. (2008) have used MLVA to study the phylogeny and the seasonal variation in V. cholerae, respectively. The initial analysis of five VNTR loci revealed the distinct populations that are present in Bangladesh and India (Ghosh et al., 2008; Stine et al., 2008; Kendall et al., 2010). Recently, Olsen et al. (2009) evaluated the usefulness of MLVA on worldwide isolates of V. cholerae based on the polymorphisms in six VNTR loci and demonstrated a high level of polymorphism among the tested strains. However, the applicability of MLVA in epidemiological investigations must be further evaluated.
In this study, we used V. cholerae O1/O139 isolate panels from a national survey to evaluate the VNTR combinations needed for the subtyping of V. cholerae for epidemiological investigations and compared them to the subtyping power of PFGE. The potential use of MLVA in phylogenetic analyses of V. cholerae O1/O139 serogroups/biotypes is also discussed.
Materials and Methods
Bacterial strains
We used 127 strains of V. cholerae isolated in China, including 89 serogroup O1 and 38 serogroup O139 strains. In addition, five reference strains were used (Supplementary Table S1; Supplementary Data are available online at
MLVA analysis
Nine VNTR markers (VC0147, VC0437, VC0500, VC1418, VC1457, VC1650, VCA0171, VCA0283, and VCA1082) were previously described (Danin-Poleg et al., 2007). The primers used for the amplification were the same as those initially reported except for VC0147, for which a new set of primers (5′-CAAACGCAGGATGAACCA-3′ and 5′-AAGAAGCCAGCGCCAATA-3′) was designed to yield bigger PCR products that allow for a better separation from the PCR products of other loci when analyzed by capillary separation. A new VNTR marker (VNTRO139-1) with a core motif of “ACCTAGA” was found from the complete genomic sequence of an O139 V. cholerae strain (isolating from a patient with ctxAB positive, unpublished data) and was also tested. VNTRO139-1 was located in the O139-specific O antigen biosynthetic gene cluster. The primers used for the amplification of VNTRO139-1 were 5′-CCACTTCACAAATATAATCAA-3′ and 5′-TTGACTGATACATGAATGGAC-3′.
The PCR reactions were prepared in a total volume of 20 μL with 10x PCR buffer, 1 unit Taq polymerase, 200 μM of each of the four dNTPs, 10 μM of each primer set, 10 ng of the DNA template, and filtered sterile water. The amplification was performed with conditions as previously described (Danin-Poleg et al., 2007). The forward primers targeting each VNTR were labeled at the 5'-end with fluorescent dyes (FAM, HEX, TAMRA, ROX). The PCR products were analyzed by capillary separation (GeneScan® ROX-500 size standard, PE Applied Biosystems) on a PE Applied Biosystems ABI Prism® 3730 instrument.
PFGE
We used the PulseNet 1-day standardized PFGE protocol for V. cholerae (Cooper et al., 2006). Optical density of cell suspensions was adjusted to 3.8–4.2 using the Densimat photometer (bioMérieux, France). V. cholerae slices were digested using 30 U per slice of NotI or SfiI (New England Biolabs, Ipswich, MA) for 4 h either at 37°C or at 50°C. Electrophoresis was performed using a CHEF-DRIII system (Bio-Rad Laboratories, Hercules, CA).
Data analysis
The data were analyzed using the BioNumerics version 5.1 software (Applied Maths, Kortrijk, Belgium). The similarity between the two PFGE patterns was expressed as a Dice coefficient (Dice, 1945). Dendrograms were clustered and constructed by using the unweighted pair group method using arithmetic averages. Nei's indices (Malorny et al., 2008) of the VNTR loci and Simpson's diversity index (Hunter and Gaston, 1988) of the subtyping methods were calculated. The formulas were as follows: Nei's index=1-∑(allele frequency)2; Simpson's diversity index (D)=1 - ∑[nj (nj - 1)]/[N(N - 1)], where nj is the number of strains belonging to the jth pattern, and N is the number of strains in the population. The D-value obtained from this equation can be applied to directly compare the subtyping methods.
Results
Level of polymorphism and discriminatory power of the VNTR loci
All loci, except VNTRO139-1, were amplified from all 52 genomes of V. cholerae strains. VNTRO139-1 was amplified only from the genomes of the O139 strains but not of the O1 strains. Nei's diversity indices of each of the nine loci were between 0 and 0.9615 (Table 1). The number of alleles of the nine loci was between 1 and 26. Locus VCA0283 had the largest number of alleles (n=26) and the highest Nei's diversity index (0.9615). Two loci (VCA0171 and VCA0283) had more than 10 alleles and Nei's diversity indices higher than 0.90; three loci (VC0147, VC0437, and VC1650) had seven alleles and Nei's diversity indices of approximately 0.70; locus VC1418 had no polymorphisms among both the O1 and O139 strains, and three loci (VC1457, VC0500, and VCA1082) had low polymorphisms among the O1 strains and no polymorphisms among the O139 strains; locus VC1457 was not polymorphic within the groups of O1 El Tor, O139, and the classical strains, but could distinguish these three groups of V. cholerae strains by giving them allele numbers of 4, 3, and 8, respectively. Eight loci had polymorphisms among the O1 strains, but only five loci had polymorphisms among the O139 strains. Overall, five loci (VC0147, VC0437, VC1650, VCA0171, and VCA0283) were highly variable, while four (VC1418, VC1457, VC0500, and VCA1082) displayed low levels of variability. VNTRO139-1 divided the 25 O139 strains into three types with Nei's diversity indices of 0.4767.
Fifty-two V. cholerae strains were distinguished into 50 MLVA profiles with a D-value of 0.9985 (Fig. 1). Only two pairs of strains were identical for all nine VNTR loci. The discriminatory power of the combination of the five highly variable loci (VC0147, VC0437, VC1650, VCA0171, and VCA0283) was equal to that observed in the case of the combination of nine loci (divided the 52 V. cholerae strains into 50 MLVA types with a D-value of 0.9985). We further compared the discriminatory power of all of the possible combinations based on five highly variable loci (Table 2). The D-values of different combinations were between 0.8944 and 0.9985. Twenty-two combinations gave D-values greater than 0.95, and only two combinations gave D-values less than 0.90. Interestingly, using only three highly variable loci (VC0147, VCA0171, and VCA0283), we showed a discriminatory power that equaled that of a combination of five highly variable loci or of all nine loci. Moreover, when the new VNTR marker, VNTRO139-1, was added, the D-value (0.9985) obtained was equivalent to that found after using the three loci for the O139 strains (divided the 25 V. cholerae serogroup O139 strains into 24 MLVA types with a D-value of 0.9967). The three-loci MLVA (VC0147, VCA0171, and VCA0283) was used for further analyses of both the O1 and the O139 strains.
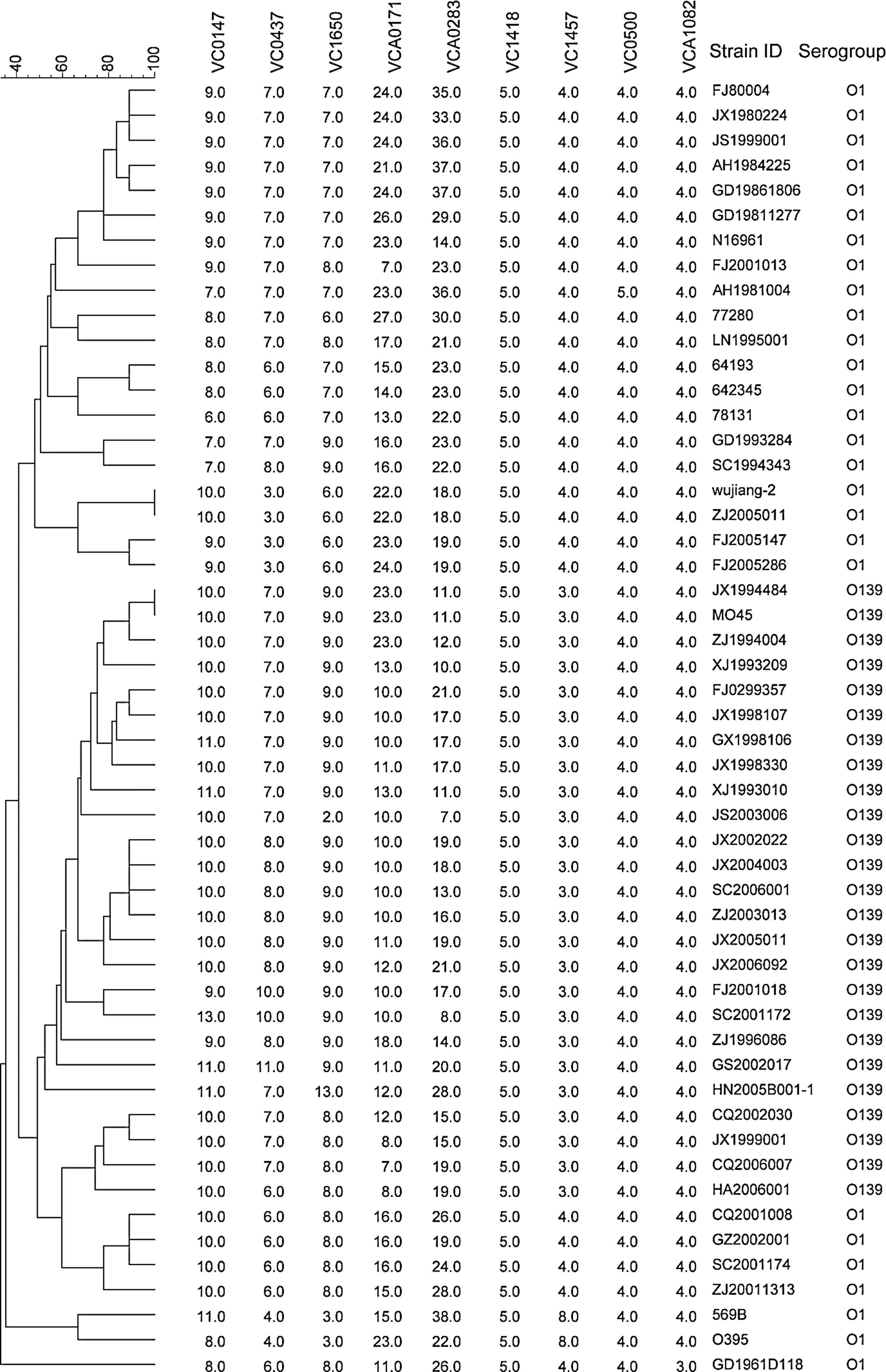
Clustering of the results of the 52 Vibrio cholerae strains with different pulsed-field gel electrophoresis patterns (differing at the genome level) by multiple-loci variable number of tandem repeats (VNTR) analysis (MLVA) using nine VNTR loci.
1, VC0147; 2, VC0437; 3, VC1650; 4, VCA0171; 5, VCA0283.
Comparison of the discriminatory power of MLVA and PFGE
The 111 epidemiologically unrelated V. cholerae strains could be distinguished at the strain level by the NotI-PFGE and SfiI-PFGE. For the 78 O1 strains, the NotI and SfiI restriction digestions produced 72 and 73 different pulsotypes with D-values of 0.9980 and 0.9973, respectively (Table 3 and Supplementary Fig. S1). For the 33 O139 strains, the NotI and SfiI restriction digestions produced 29 and 23 different pulsotypes with D-values of 0.9905 and 0.9261, respectively (Table 3 and Supplementary Fig. S2). D-values of 1.0000 and 0.9981 were obtained by combining using of NotI- and SfiI-PFGE for 78 O1 and 33 O139 strains, respectively. By three-loci MLVA, 70 types with a D-value of 0.9970 were found in the 78 O1 strains; in the 33 O139 strains, 28 types with a D-value of 0.9848 were found. For the O1 strains, both methods yielded a D-value>0.99. For the O139 strains, MLVA yielded a D-value similar to that found in NotI-PFGE and slightly higher than that observed in SfiI-PFGE.
The pattern frequency distribution in the PFGE and the MLVA analyses using the strains listed in strain panel 2 are shown in Figure 2. For the O1 strains (Fig. 2A), 66 and 70 patterns, which accounted for 91.7% (66/72) and 95.9% (70/73) of the NotI-PFGE and the SfiI-PFGE subtyping, respectively, contained only one strain in each. By combining using of NotI- and SfiI-PFGE, all strains had a single pattern. Sixty-three types, accounting for 90.0% (63/70) of the MLVA subtyping, contained one strain in each. These patterns accounted for 80.8% (63/78) of the strains tested. These results indicated that MLVA had a discriminatory power for the subtyping of the O1 strains similar to that of PFGE.

Frequency distribution of patterns obtained by pulsed-field gel electrophoresis (PFGE) (NotI and SfiI individually and combined) and three-loci multiple-loci variable number of tandem repeats (VNTR) analysis (MLVA) (VC0147, VCA0171, and VCA0283). The figures on the x-axis indicate the number of strains in each of the patterns. Y-axis indicates the sum of the patterns.
For the O139 strains (Fig. 2B), 27 patterns, which amassed to 90.0% (27/30) of the NotI-PFGE subtyping, contained individually only one strain. Moreover, these patterns contained 81.8% (27/33) of the test strains. Twenty-one patterns, covering 91.3% (21/23) of the SfiI-PFGE subtyping, contained one strain per pattern. The patterns derived from the SfiI-PFGE had 63.6% (21/33) of the test strains. By combining using of NotI- and SfiI-PFGE, 31 patterns (93.9%) contained only one strain in each. For the MLVA subtyping, 25 subtypes, accounting for 89.3% (25/28), contained one strain per subtype. These subtypes accounted for 75.8% (25/33) of the test strains. One pattern of the SfiI-PFGE contained nine strains, which indicates that this method had a lower discriminatory power for the subtyping of the O139 strains than that of NotI-PFGE and MLVA.
Comparison of the epidemiological concordance of PFGE and MLVA
The analysis of the epidemiologically related strains demonstrated that most of the strains within each outbreak had identical or very similar patterns (subtype) when analyzed by either PFGE or MLVA (Fig. 3). Both methods were able to group strains from all V. cholerae outbreaks into clusters representing the different pulsotypes except for outbreak1 and outbreak2 when analyzed by SfiI-PFGE analysis. SfiI-PFGE could not distinguish outbreak1 and outbreak2. It was also observed that PFGE clustered some outbreak strains into different patterns, but MLVA clustered the outbreak strains of each outbreak quite well (Fig. 3). Furthermore, by combining using of NotI- and SfiI-PFGE, there were only two outbreaks (outbreaks 2 and 4) completely clustered together of each.

Clustering of the 26 Vibrio cholerae strains isolated from seven outbreaks by using pulsed-field gel electrophoresis (PFGE) (NotI and SfiI individually and combined) and three-loci multiple-loci variable number of tandem repeats (VNTR) analysis (MLVA) (VC0147, VCA0171, and VCA0283). Both the serogroups and the serial number of the outbreak are listed on the right of each strain.
The ability for phylogenetic analysis of 3-, 5-, 6-, and 9-loci MLVA for V. cholerae
Although the three-loci MLVA could divide the 72 strains into the different subtypes at the strain level, these three loci (VC0147, VCA0171, and VCA0283) could not cluster the O1 El Tor ctxAB-positive strains, the O139 ctxAB-positive strains, and the classical strains in each group (Fig. 4A). Five loci (VC0147, VC0437, VC1650, VCA0171, and VCA0283) could cluster the three classical strains together, but the ctxAB-positive O1 and O139 strains remained dispersed in the phylogenetic tree (Fig. 4B). When six loci (VC0147, VC0437, VC1457, VC1650, VCA0171, and VCA0283) were used, the O1 El Tor ctxAB-positive, the O139 ctxAB-positive, and the classical strains were clustered in each group (Fig. 4C), while the ctxAB-negative strains were dispersed in the phylogenetic tree, similar to what was found when all nine loci were used (Fig. 4D). However, except for the serogroups, biotypes, and ctxAB, there was no further cluster feature associated with other characteristics within the ctxAB-positive O1 and O139 strains, such as the year of isolation, the regions the strains are from, or the sources of isolation.

Minimum spanning tree analysis of 72 Vibrio cholerae strains by multiple-loci variable number of tandem repeats (VNTR) analysis (MLVA) using different combinations of VNTR loci.
Discussion
In 2007, Danin-Poleg et al. found that VNTRs were widespread and randomly distributed in the two chromosomes of the V. cholerae genome (Danin-Poleg et al., 2007). Subsequent studies showed that MLVA using different VNTRs was efficient for V. cholerae subtyping and could be used as a tool for epidemiological studies of cholera (Stine et al., 2008; Kendall et al., 2010). In this article, based on a panel of 52 ctxAB-positive V. cholerae strains, we showed that five loci had significant levels of polymorphism, while four other loci had a much lower discriminatory power. In our study, seven loci gave lower D-values and the VCA0283 locus gave a significantly higher D-value than the D-values seen in Danin-Poleg's study. These differences may be caused by the different panel of strains used. In our study, only the ctxAB-positive O1 and O139 strains but not the non-O1/non-O139 strains were selected as a panel to evaluate the discriminatory power of VNTRs.
Based on the 52-strains panel, three-loci MLVA (including VC0147, VCA0171, and VCA0283) showed enough subtyping power compared to various combinations of other loci. This phenomenon was further demonstrated with epidemiologically unrelated strains. Similar subtyping capacities of the three-loci MLVA were observed when compared with PFGE. An optimal molecular subtyping method used during outbreak investigations requires not only a good discriminatory power, but also a high epidemiological concordance. Epidemiological concordance refers to the ability to link epidemiological related isolates, especially those from the same outbreak. In this study, it was interesting to see that PFGE separated some outbreak strains into different but very closely related patterns, whereas three-loci MLVA grouped together strains of each outbreak, thus showing a higher epidemiological concordance. Although strains may undergo slight variation during an outbreak, it appears that the three-loci MLVA developed in our study may reflect the relative stability of the strain genomes during an outbreak and may facilitate case definition during an epidemiological investigation. Our findings show the potential significance of this MLVA protocol, despite the fact that a broad validation of this protocol is still needed.
MLVA methods using a handful of highly variable loci have also been suggested in previous studies. Bhowmick et al. (2010) evaluated one locus (VCA0171, named VcA VNTR in that study) as a method for strain subtyping and phylogeny studies of V. cholerae, and considerable potential of VCA0171 VNTR analysis as a tool for the subtyping of V. cholerae was emphasized. In a recent study, analysis using the two highly variable loci—VCA0171 and VCA0283—produced D-values comparable with those obtained using six loci (VC0147, VC0437, VC1457, VC1650, VCA0171, and VCA0283), which suggests that there is a reduced need for the use of more markers during outbreak situations, thereby saving both time and resources (Lam et al., 2012).
In the current study, we also found that MLVA based on six loci could be used to cluster the O1 El Tor ctxAB-positive, the O139 ctxAB-positive, and the classical strains in each group, while the ctxAB-negative O1 and O139 strains were dispersed in the phylogenetic tree. This population structure of the V. cholerae is similar to that obtained by a DNA array-based comparative genomic hybridization (Pang et al., 2007). Locus VC1457 had no polymorphisms within the tested groups of the O1 El Tor, the O1 classical, and the O139 strains, but it could still distinguish these three groups of V. cholerae strains; this locus is important to build an accurate minimum spanning tree. In a recent study, the researchers concluded that MLVA is best used in conjunction with single nucleotide polymorphism subtyping to most accurately determine the evolutionary relationships among the seventh pandemic V. cholerae isolates and for longer-term epidemiological subtyping (Lam et al., 2012). Our results further indicate that these six loci also have serogroup- and biotype-specific characteristics and can also assist in distinguishing toxigenic strains from environmental nontoxigenic ones.
Rapid molecular subtyping methods with appropriately high discriminatory power are continuously in demand for the surveillance and quick response during an outbreak. In this study, both MLVA and PFGE had the ability to subtype all of the V. cholerae strains tested and could be used as subtyping methods for V. cholerae with a generally accepted 5% probability of type I errors. We suggest three loci (VC0147, VCA0171, and VCA0283) for V. cholerae MLVA subtyping for the surveillance of cholera. This approach would be particularly useful in the case of an outbreak. Three-loci MLVA can be performed by one multiplex PCR followed by one capillary electrophoresis. Finally, the three-loci MLVA technique is also cost effective.
Conclusions
We have shown that the high discriminatory power of the three-loci MLVA (VC0147, VCA0171, and VCA0283) for V. cholerae subtyping equaled that of PFGE. The three-loci MLVA could be solely used as a subtyping method during cholera outbreak investigations. Six loci (VC0147, VC0437, VC1457, VC1650, VCA0171, and VCA0283) can be used when studying the phylogenetic relationships and the population structure of the V. cholerae O1/O139 strains, and little to no benefit was obtained using nine loci over the six in this study.
Footnotes
Acknowledgments
This study was supported by the Priority Project on Infectious Disease Control and Prevention (2012ZX10004215) from Ministry of Health, China and National Basic Research Priorities Program (2009CB522604) from the Ministry of Scientific Technology, China.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.