
Abstract
A gene-specific microsphere suspension array coupled with 15-plex polymerase chain reaction (PCR) was developed to screen bacterial samples rapidly for 10 strains of bacteria: Shigella spp. (S. flexneri, S. dysenteriae, and S. sonnei), Staphylococcus aureus, Vibrio cholerae (serology O1 and O139), Legionella pneumophila, and Clostridium botulinum (types A, B, and E). Fifteen sets of highly validated primers were chosen to amplify target genes simultaneously. Corresponding oligonucleotide probes directly conjugated with microsphere sets were used to specifically identify PCR amplicons. Sensitivity tests revealed that the array coupled with single PCR was able to detect purified genomic DNA at concentrations as low as 10 copies/μL, while the multiplex detection limit was 10–104 copies/μL. The assay was validated using water samples artificially spiked with S. aureus and S. dysenteriae, as well as water specimens from swimming pools previously identified to contain S. aureus.
Introduction
T
Since its invention in 1985, the polymerase chain reaction (PCR) has offered significant advantages in pathogen detection and identification. PCR has a theoretical detection limit of one single pathogenic bacterium, and it can identify microbes that cannot be identified by either culture or immunological methods (Batt, 2007). Various PCR-based molecular methods have been developed, such as real-time PCR and oligonucleotide microarrays. Recently, the suspension arrays or liquid chips, a brand-new array technology developed by Luminex Corp., have a robust capability for DNA assays, immunoassays, receptor-ligand assays, and enzyme assays (Kim et al., 2009; She et al., 2010; Chikkaveeraiah et al., 2011). This suspension arrays capitalizes on the strengths of flow cytometry. Multiplexed analysis is made by using a set of fluorescent latex microspheres that can be read by red and orange fluorescence (Fulton et al., 1997; Kettman et al., 1998).
Shigella spp., Staphylococcus aureus, Vibrio cholerae, Legionella pneumophila, and Clostridium botulinum are the common bacterial pathogens implicated in foodborne or waterborne illnesses. In this study, our final objective was to develop a multiplex detection system for these pathogens and thereby validate the utility of microsphere-based suspension arrays for detecting real samples for pathogen.
Materials and Methods
Bacterial strains and DNA extraction
The following bacterial strains were used as standard strains in this study: Shigella flexneri CMCC (National Center For Medical Culture Collections, Beijing, China) 51062; S. dysenteriae CMCC 51197; S. boydii CMCC 51265; S. sonnei CMCC 51334; Staphylococcus aureus CMCC 26003, and ATCC (American Type Culture Collection, Rockville, MD) 6538; Vibrio cholerae O1 IME (Beijing Institute of Microbiology and Epidemiology) 860084; V. cholerae O139 vaccine strain used; Clostridium botulinum type A, strain 62A; C. botulinum type B, strain 621; and C. botulinum type E, strain 619. One clinical strain of Legionella pneumophila was kindly provided by Dr. Shouyi Chen. More than 30 strains spanning 14 genera (listed in Table 1) were used to determine the specificity of the detection method. Total genomic DNA was extracted from laboratory cultures by Bacteria DNA kits (Qiagen, Dusseldorf, Germany) according to the manufacturer's instructions.
ATCC, American Type Culture Collection; CMCC, China Medical Culture Collection Center.
Design of primers and probes
The target genes selected for PCR amplification are unique for each pathogen, and in previous studies they were found to be highly conserved within the species. They included: ipaH (Buysse et al., 1987) for Shigella; ial (Islam and Lindberg, 1992) for virulent Shigella; wzy (Houng et al., 1997; Farfan et al., 2010) for Shigella sonnei; rfc (Houng et al., 1997) for Shigella flexneri; rfbx (Houng et al., 1997) for Shigella dysenteriae; ctx (Shirai et al., 1991; Chapela et al., 2010) for Vibrio cholerae; rfbE (Stroeher et al., 1992; Huang et al., 2009) for Vibrio cholerae O1; LPS (Stroeher et al., 1997; Huang et al., 2009) for Vibrio cholerae O139; nuc (Brakstad et al., 1992) and scpA (Golonka et al., 2004) for Staphylococcus aureus; mip (Engleberg et al., 1989; Wolter et al., 2008) for Legionella pneumophila; bont/A, B, and E (Szabo et al., 1993) for Clostridium botulinum type A, B, and E, respectively.
Fourteen specific genes in the multiplex PCR approach were selected as targets for each pathogen. Primer sets were designed from target regions with Primer Premier Software (version 5.0). Primer sets had annealing temperatures of 55°C–58°C, GC contents of 40%–50%, and lengths of 19–23 bases. The primers were validated for specificity by BLAST analysis available through the NCBI website. For suspension array assay, we chose primer sets that generate 100–400 bp amplicons, and one of the primer sets was synthesized with a biotin modification at the 5' end. The oligonucleotide probes were chosen from the amplicons with annealing temperatures of 55°C–58°C. An amino-(CH2)12 modifier was incorporated on the 5' end of each probe to reduce steric hindrance from microspheres. The probes were validated for high specificity by BLAST. Both probes and primers were synthesized by TaKaRa (Dalian, Liaoning, China). In addition, a common primer set and probe from the 16S rDNA conserved region were used as an internal control (IC) for evaluating the efficiency of PCR and the hybridization system.
Single and multiplex PCR reactions
Each PCR amplification was performed in a total volume of 20 μL with the T-personal thermal cycler (Biometra, Goettingen, Germany). The reaction mixture consisted of 1.5 U of TaKaRa Taq™ polymerase, 0.05 U UNG (Fermentas, USA), 1×TaKaRa Taq™ PCR buffer, 0.2 mM dATP, dCTP, and dGTP, 0.1 mM dTTP and dUTP, 1–10 ng purified genomic DNA, 15 oligonucleotide primer sets ranging from 20 to 160 nM, and enough ddH2O to achieve a final reaction volume of 20 μL. Biotin-labeled primers and nonlabeled primers were used in a 1:1 ratio to produce symmetric amplification of PCR products (symmetric PCR), or in a 5:2 ratio to promote asymmetric amplification of the probe complement strand (asymmetric PCR). The amplification procedures included an initial 5-min incubation at 37°C, 94°C for 3 min, 35 cycles of amplification with denaturation at 94°C for 30 s, 53°C for 40 s, 72°C for 40 s, and final extension at 72°C for 3 min. A PCR blank tube (all PCR reagents minus the DNA template) was used to monitor the potential carryover contamination in each assay.
Coupling of probes to microspheres
Each specific probe was covalently coupled to 5.6-μm polystyrene carboxylated microspheres (Luminex, Austin, TX) using a carbodiimide coupling procedure according to studies (Dunbar, 2006; Baums et al., 2007) with some modifications. Briefly, 200 μL (about 2.5×107 microspheres) of microsphere solution was transferred to a 2.0-mL microfuge tube. The microspheres were pelleted by centrifugation at 12,000×g for 1 min and the supernatant was discarded. The microspheres were resuspended in 50 μL of 0.1 M MES (2[N-morpholino] ethanesulfonic acid, Sigma, Munich, Germany), pH 4.5. Amino-modified probe (0.2 nmol) was added and the solution was vortexed. A 1-μg/μL solution of EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride; Pierce, Rockford, IL) was freshly made, and 2.5 μL was added to the microsphere solution and vortexed, followed by incubated for 30 min. Another fresh batch of EDC solution was made, and the EDC addition and incubation steps were repeated. Coupled microspheres were washed with 1 mL of 0.02% Tween 20, followed by 0.1% sodium dodecyl sulfate, and were resuspended in 100 μL of Tris-EDTA (TE) buffer. The suspension was enumerated in a cell-counting chamber and stored in the dark at 4°Cuntil use.
Suspension array assay
The biotin-labeled PCR products were hybridized to the probe-coupled microspheres (Hadd et al., 2004). The reaction mixture consisted of 1×TMAC (tetramethylammonium chloride, Sigma, Munich, Germany) hybridization buffer (4.5 M TMAC, 0.12% Sarkosyl [N-lauroylsarcosine], 7.5 mM Tris-HCl, 6 mM EDTA, pH 8.0), 5000 microspheres of each set, 5 μL of biotinylated amplicons, and 1×TE added to achieve a final reaction volume of 50 μL. The hybridization program included denaturation at 94°C for 3 min, and hybridization at 52°C for more than 15 min.
After hybridization, the reaction solution was transferred to a 96-well filter plate (Millipore, Darmstadt, Germany) and the microspheres were pelleted by vacuum filtration. The hybridizing amplicons were resuspended in 75 μL of 1×TMAC solution containing 2 μg/μL of streptavidin-R-phycoerythrin (Invitrogen, Foster, CA). The plate was incubated at 52°C for 10 min and read on a heated plate at the same temperature on the Luminex200 platform (Luminex, Austin, TX). For each probe, 100 microspheres were analyzed, representing 100 replicate measurements. The median fluorescence intensity (MFI) measurements were calculated by the Luminex digital signal processor and proprietary software. The MFI was corrected for background (F-F0, where F0 was the background signal determined from the fluorescence measurement of a PCR blank sample). A positive suspension array response has been defined as a signal twice the background after the background has been subtracted.
Sensitivity and specificity of the suspension array assay
The sensitivity of the suspension array assay was determined in singleplex and multiplex formats with purified genomic DNA of S. dysenteriae, V. cholerae O1, and S. aureus strains using the conditions described. The genomic DNA was quantitated spectrophotometrically by Microspectrophotometer and then diluted 10-fold. The final concentration ranged from 100 copies/μL to 105 copies/μL. The specificity was determined with 1–10 ng of genomic DNA from the strains described in singleplex and multiplex PCR format array assays.
Suspension array detection of Shigella and S. aureus in simulated water
A liquid culture of S. dysenteriae and S. aureus at known concentrations based on a quantitative culture method was used to assess the detection limit of the multiplex PCR suspension assay for environmental specimens. Cells were inoculated into 100 mL water at concentrations ranging from 0 to 107 CFU/mL. Twenty milliliters of each water sample was filtered through a 0.22-μm, 25-mm cellulose acetate membrane filter (Millipore, Darmstadt, Germany), which was then rinsed with 10 mL of phosphate buffered saline. The membranes were processed directly using MicroDNA Kits (Qiagen, Dusseldorf, Germany) according to the manufacturer's instructions. The final eluted volume was 40 μL in ddH2O. Single and multiplex PCR were performed using 10 μL of the eluate as the PCR template.
Suspension array detection of real water samples
We collected the contaminated water from a swimming pool with previous real-time PCR results of a DaAn Gene Detection Kit for Staphylococcus aureus (PCR-fluorescence probing). DNA was extracted using the methods described above. The suspension array with multiplex PCR was used to screen pathogens from real samples.
Results
Suspension array assay
The targeted genes, selected oligonucleotide primer sets, and probes used for the detection of each pathogen are listed in Table 2. By optimizing two key factors of annealing temperature and the Taq™ polymerase concentration, a 15-plex PCR reaction was finally established. The ideal annealing temperature was 53°C and adopted 1.5 U Taq in a 20-μL reaction volume. We got a high MFI for an amplification of the probe complement strand with a ratio of 5:2 (biotin-labeled primers: nonlabeled primers) by using an asymmetric PCR method (data not shown). The conditions of hybridization were determined at 52°C for more than 15 min.
Biotin modification at the 5' end.
Using the multiplex detection conditions described above, we successfully amplified 15 targeted gene segments (Fig. 1A). In each tube, we detected S. flexneri, S. dysenteriae, S. boydii, S. sonnei, V. cholerae O1, V. cholerae O139, L. pneumophila, C. botulinum type A, B, and E, and S. aureus, respectively (Fig. 1B).
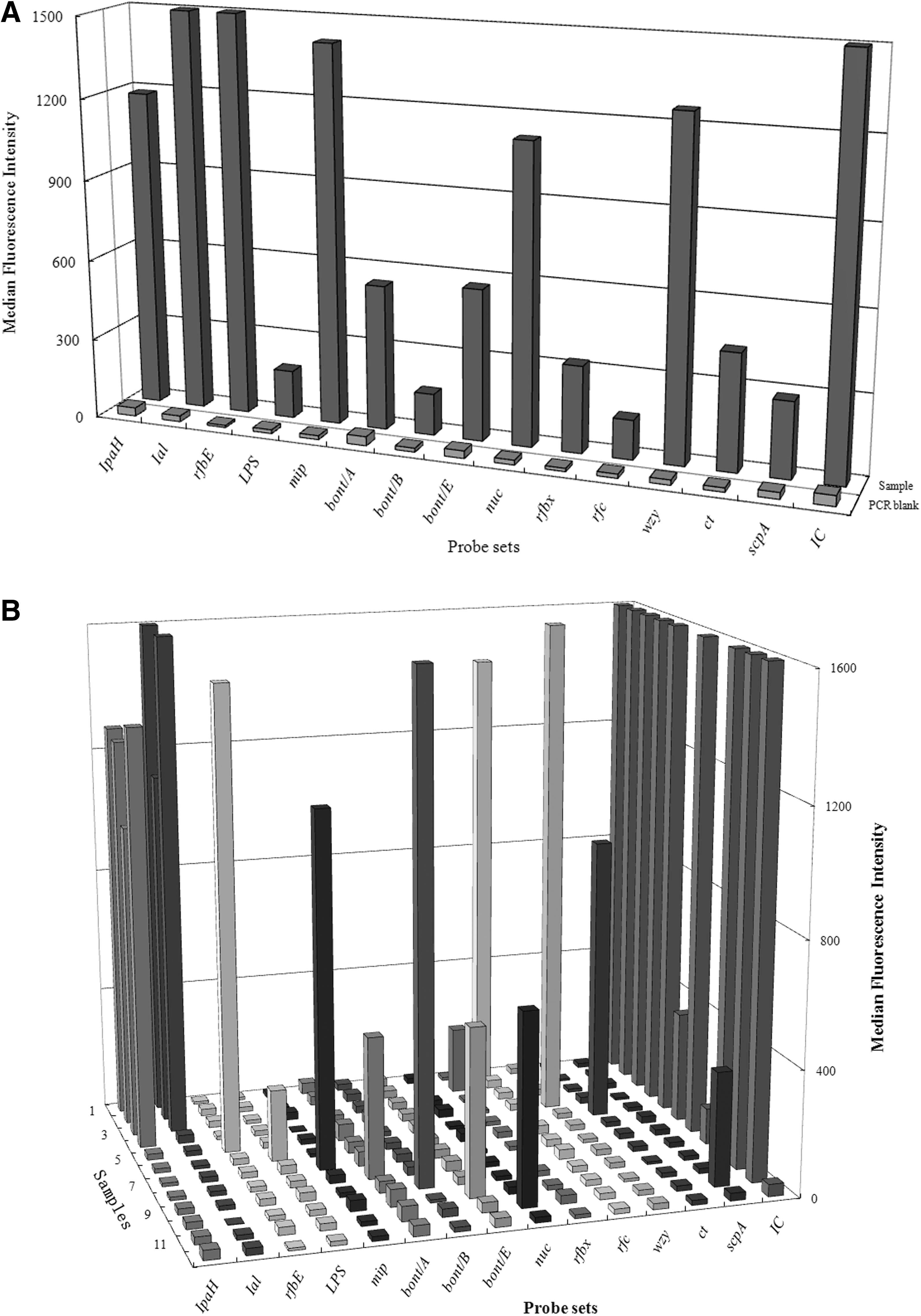
Results of suspension array multiplex detection pathogens with a pool of DNA template
Sensitivity and specificity of the suspension array
The sensitivity of the array assay was determined with genomic DNA extracted from cultured S. dysenteriae, V. cholerae O1, and S. aureus. The amount of genomic DNA needed for detection by single PCR corresponded to 100 cells for IpaH and Ial of S. dysenteriae, 100 cells for rfbE, 101 cells for ctx of V. cholerae O1, and 101 cells for nuc and scpA of S. aureus. For detection by multiplex PCR, the DNA required corresponded to 103 cells for IpaH, 104 cells for Ial of S. dysenteriae, 102 cells for rfbE, 104 cells for ctx of V. cholerae O1, 104 cells for nuc and scpA of S. aureus (Table 3). Independent analyses were run three times to test the detection level.
The amount of genomic DNA of detection limitation corresponding to one cell for IpaH and Ial of S. dysenteriae, one cell for rfbE, and 10 cells for ctx of V. cholerae O1, 10 cells for nuc and scpA of S. aureus by single PCR (sPCR), while 1000 cells for IpaH and 10,000 cells for Ial of S. dysenteriae, 100 cells for rfbE and 10,000 cells for cxt of V. cholerae O1, 10,000 cells for nuc and scpA of S. aureus by multiplex PCR (mPCR).
The specificity was tested using genomic DNA from all of the strains involved in this study. Suspension array has the advantage of the double specificity provided by the primer set and probe. All target bacteria gave positive signals, while 30 strains of nontarget bacteria (listed in Table 1) were negative, indicating 100% specificity.
Suspension array detection of S. dysenteriae and S. aureus in simulated water
The utility of suspension arrays to detect pathogens in simulated environmental samples was tested with 10-fold dilutions of S. dysenteriae and S. aureus cells. The suspension array assay coupled with singleplex PCR was able to detect ipaH, ial, nuc, and scpA at 2×103 CFU/100 mL, 2×102 CFU/100 mL, 2×103 CFU/100 mL, and 2×102 CFU/100 mL, respectively. In contrast, assays coupled with multiplex PCR were capable of detecting ipaH, ial, nuc, and scpA at 2×104 CFU/100 mL, 2×105 CFU/100 mL, 2×105 CFU/100 mL, and 2×105 CFU/100 mL, respectively (S. dysenteriae results are shown in Table 4). The high MFI of the IC demonstrated successful amplification and hybridization, while the low MFI of the PCR blank indicated the absence of carryover contamination.
One hundred milliliter water samples were seeded with serially diluted S. dysenteriae cells corresponding to 2×100 cells, 2×101 cells, 2×102 cells, 2×103 cells, 2×104 cells, 2×105 cells, and 2×106 cells followed by DNA extraction for single assays and multiplex assays.
Suspension array detection of real water samples
The higher MFI of the nuc and scpA of S. aureus was detected in amplicons from a contaminated water DNA, while the same low MFI was showed on those probes from Shigella spp., V. cholerae, C. botulinum, and L. pneumophila as the PCR blank (Table 5). It inferred that swimming water could be contaminated by S. aureus. This result agreed with real-time PCR results of the DaAn Gene Detection Kit for Staphylococcus aureus (PCR-Fluorescence Probing).
A, B, C were parallel samples.
Discussion
Many multiplex detection systems based on PCR have been developed for pathogen profiling; however, because of some inherent drawbacks, their practical application remains a distant goal. In this study, we have extended the utility of the suspension array coupled with 15-plex PCR to simultaneously screen for waterborne pathogens in the single tube. Actually, three basic strategies have been introduced for pathogens detection based on suspension arrays: multiplex PCR amplification, universal primer amplification, and direct hybridization to RNA. Multiplex PCR has the advantage of the double specificity provided by the primer set and probe. In addition, multiplex capabilities have been significantly extended to much more multiplex than 15-plex such as 18-plex (Lee et al., 2007), 25-plex (Han et al., 2006), and even 90-plex PCR coupled with microarrays (Vora et al., 2005). Unfortunately, as the number of primer sets in a reaction is increased, there is a trend toward reduced sensitivity of the multiplex PCR. Comparing in single and 15-plex formats, the sensitivity of 15-plex detection decreased 10–1000 times compared to singleplex detection. A similar result was also published by Wilson (Wilson et al., 2005). Our results also indicate that sensitivity is improved by increasing Taq polymerase concentration. For example, we adopted 1.5 U Taq in a 20-μL reaction volume, which tripled the concentration recommended by the manufacturer. It was reported that 15 U of Taq polymerase was also employed in 50-μL volumes to perform 90-plex PCR reactions for enhancing sensitivity, but false positives will be increased because of more manipulations of PCR products (Vora et al., 2005). Although universal primers are capable of high sensitivity and can detect all bacteria using one primer pair from the 16S/23S rDNA (Spiro and Lowe, 2002; Diaz and Fell, 2004), they have the disadvantage of cross-reaction at the species level and between some genera because of a high degree of genetic similarity (Maynard et al., 2005). Furthermore, bacterial diversity in environmental samples may decrease the efficiency of amplification of interesting bacteria. In addition, PCR-based methods are poorly suited to determine pathogen quantities and viability. Direct hybridization of RNA provides the least bias in gene detection (Chandler and Jarrell, 2004) but the lowest level of assay sensitivity.
Any PCR-based detection system, while potentially rapid, sensitive, and specific, must still confront a number of inherent drawbacks. Fortunately, when a suspension array is coupled with multiplex PCR, some of the drawbacks may be eliminated. Conventional multiplex PCR followed by agarose gel electrophoresis usually does not exceed 6-plex because it is difficult for agarose gels to separate more than six fragments, and to allow for interpretation of potential nonspecific products. In this article, we describe a suspension array coupled with 15-plex PCR for pathogens detection; theoretically, the suspension array can identify up to 100 agents. Especially, nonspecific amplification is also not a big problem for array method because only specific amplicons will hybridize to the oligonucleotide probes on the microspheres. Suspension arrays provide another approach to enhance the identification of PCR products. In addition, hybridizations were performed in TMAC, in which the hybridization condition depends on the sequence length and not on the base composition, thereby reducing hybridization bias based on oligonucleotide variations (Wood et al., 1985). Moreover, the surface chemistry on microspheres appears to be more stable, which enables hybridization reactions to be performed at ideal temperatures and allows simple process protocols to be used (Spiro et al., 2000).
Because of the potentially extreme sensitivity and powerful amplification of PCR, false-positive results due to carryover contamination have become a significant challenge for PCR-based detection systems. The same concept applies to microarrays (Warsen et al., 2004). Furthermore, we usually have to manipulate PCR products in the assays, which increases the occurrence of false positives. Thus, strategies must be adopted to control the potential carryover contamination for any PCR-based method. Herein, we used UNG to overcome this problem (Hartley and Rashtchian, 1993). In addition, each assay incorporated a PCR blank to monitor potential carryover contamination.
Conclusions
Suspension array is a rapid and effective platform for the simultaneous, multiplexed detection of pathogenic organisms, and in a single tube probes specific for foodborne and clinically relevant organisms could be easily added. It will become a routine diagnostic tool in the near future if the following questions are addressed: What is a suitable PCR approach to match the suspension array? What is the specificity and sensitivity of the detection system? Ultimately, validation and high-throughput sample preparation will continue to be significant challenges for pathogen detection using suspension array.
Footnotes
Acknowledgments
The authors gratefully acknowledge the financial assistance provided by the China National 863 high-technology project (2006AA06Z414).
Disclosure Statement
No competing financial interests exist.