
Abstract
Listeria monocytogenes, an emerging foodborne pathogen, can cause in the population at risk severe infections that are associated with high case fatality rates. A total of 93 L. monocytogenes strains isolated from different patients in Switzerland from July 2011 to September 2013 were further characterized. Septicemia was reported for 74.2% of the patients, meningitis for 10.8%, and abortion for 3.2%. The majority of the strains belonged to serotype 1/2a (n=58) followed by serotype 4b (n=28), 1/2b (n=5), and 1/2c (n=2). The strains represented 35 multilocus sequence typing sequence types, 8 of which were designated for the first time. Sequence analysis of the inlA gene in the 35 sequence types showed that most of the strains encoded full-length proteins. Screening for Listeriolysin S showed the presence of this virulence factor in 29 of the 33 genetic lineage I strains. By using ApaI and AscI for pulsed-field gel electrophoresis, most strains showed distinguishable patterns.
Introduction
L
To gain insight into the relationships and virulence-associated characteristics of strains occurring in Switzerland, the aim of the present study was to characterize all clinical L. monocytogenes strains collected by the Swiss National Reference Laboratory from 2011 through 2013 by phenotyping and genotyping methods and to compare these results with data from other countries in Europe and the United States.
Materials and Methods
Strains
The 93 strains were isolated from July 2011 to September 2013 from human patients with a severe illness and a serious suspicion of infection with L. monocytogenes. Due to mandatory notification and the obligation of laboratories to forward human L. monocytogenes strains to the National Reference Laboratory, these strains are considered representative for the period screened.
Lineage polymerase chain reaction (PCR)
Lineage-specific PCR assays were performed using the primers and protocol described by Ward et al. (2008). Reactions were carried out in a 20-μL volume that contained 1x GoTaq Green Master Mix (Promega, Madison, WI).
Serotyping
Serotyping was performed using the commercial set of Listeria O-factor and H-factor antisera from Denka Seiken (Pharma Consulting, Burgdorf, Switzerland).
Antimicrobial susceptibility testing
Strains were tested for antimicrobial susceptibility by the disc-diffusion method according to the protocols of the Clinical and Laboratory Standards Institute (Anonymous, 2013b). The antibiotics (Becton Dickinson, Sparks, MD) tested were the following: ciprofloxacin 5 μg (CIP5), rifampicin 5 μg (RA5), gentamicin 10 μg (GM10), ampicillin 10 μg (AM10), erythromycin 15 μg (E15), vancomycin 30 μg (VA30), clindamycin 2 μg (CC2), tetracycline 30 μg (TE30), and sulfamethoxazole 23.75 μg/trimethoprim 1.25 μg (SXT23.75/1.25).
PFGE subtyping and cluster analysis
Pulsed-field gel electrophoresis (PFGE) analysis of the strains was done in accordance with the CDC PulseNet protocol (
For pattern evaluation, BioNumerics v6.5 (Applied Maths, Sint-Martens-Latem, Belgium) was used. Pairwise similarities between the ApaI and AscI PFGE patterns were calculated using the JACCARD similarity coefficient. Cluster analysis was performed on the unweighted pair-group method with averages. Tolerance and optimization parameters were set to 1.5% each. Salmonella serovar Braenderup strain H9812 (ATCC BAA 664) digested with XbaI (100 U/plug; Roche Diagnostics) was used as size standard. For each restriction enzyme, the different patterns were grouped into pulsotypes based on a cut-off value of 95% pattern similarity.
MLST
Multilocus sequence typing (MLST) was performed as described by Ragon et al. (2008) with the following modifications: Amplifications were performed in 50 μL volume using the HotStarTaq Master Mix (Qiagen) and 50 ng of genomic DNA. Following cycling, conditions were determined for amplification of the bglA, cat, and ldh loci: 94°C for 4 min, followed by 42 cycles of 94°C for 30 s, 45°C for 30 s, and 72°C for 2 min; for the abcZ, dapE, dat, and lhkA loci: 94°C for 4 min followed by 35 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 2 min. Amplifications were finalized with a 10-min 72°C step. Sequencing of the amplicons was performed employing the universal primers described by Ragon et al. (2008). Alleles and sequence types (STs) are publicly available at
Internalin A gene (inlA) sequence profiling
Internalin A profiling was performed on one representative strain of each ST identified in this study using PCR primers described previously (Orsi et al., 2007). PCR amplifications were performed using the Phusion High Fidelity Taq Polymerase and conditions recommended by the supplier (Fermentas, St. Leon-Rot, Germany). Amplicons were sequenced commercially by Microsynth (Balgach, Switzerland) and analyzed for mutations with CLC Main Workbench 6.6.1.
Further strain characterization
The presence of Listeriolysin S was determined as described by Cotter et al. (2008). PCR reactions were performed in 50-μL volume, using 1 x GoTaq Green Master Mix (Promega). PCR amplifications for the 50-kb Listeria genomic island (LGI1) (Gilmour et al., 2010) were performed on serotype 1/2a strains using primers and PCR conditions described by Kovacevic et al. (2012). Reactions were carried out in 50-μl volume that contained 1 x GoTaq Green Master Mix (Promega). PCRs to determine the presence of the stress survival islet (a five-gene region consisting of lmo0444-lmo0448) were performed as described by Ryan et al. (2010). The Phusion High Fidelity Taq Polymerase was used for the PCR amplifications in accordance with the supplier's recommendations (Fermentas).
Results and Discussion
Origin of the strains
Most of the 93 human strains included in this study were isolated from patients with severe illness between July 2011 and September 2013. The origins of the strains for these patients are summarized in Table 1. Septicemia was reported for 74.2% of the patients, meningitis for 10.8%, and abortion for 3.2%. Furthermore, gastroenteritis was reported for 3.2% of the patients, colpitis for 1.1%, and inflammation of the knee joint for 1.1%. For 6.5% of the patients no medical history data were available. Five patients (5.4%) were aged <10 years, 60 (64.5%) were aged >60 years, and 24 (25.8%) patients were aged between 10 and 60 years. For four patients no data were available. Of the reported cases, 50.5% were female and 49.5% were male.
B, blood; L, cerebrospinal fluid (liquor); P, placenta; O, other/no information.
LLS, Listeriolysin S;+, positive; –, negative.
SSI-1, stress survival islet;+, positive, –, negative; DA, SSI-1 polymerase chain reaction (PCR) amplicons obtained differ from those expected in either positive or negative strains; NA, the SSI-1 PCR did not give any amplicon.
Only clonal complexes/singletons containing 4 strains or more are represented; all other CC (CC3, 5, 59, 224, 517) are summarized as “different CC.”
Only clonal complexes/singletons containing four strains or more are represented; all other CC (CC2, 54) are summarized as “different CC.”
Only clonal complexes/singletons containing 4 strains or more are represented; all other CC (CC7, 9, 14, 16, 26, 29, 36, 37, 101, 121, 155, 199, 204, 207, 394, 398, 412, 504, 671) are summarized as “different CC.”
Only clonal complexes/singletons containing four strains or more are represented; all other CC (CC9) are summarized as “different CC.”
Lineages and serotypes
Lineage determination was achieved based on the MLST data and confirmed by lineage specific PCR. Strains belonged either to lineage II (60 strains) or to lineage I (33 strains).
The majority of the strains belonged to serotype 1/2a (n=58) followed by serotype 4b (n=28), 1/2b (n=5), and 1/2c (n=2) (Table 1). Serovar 1/2a was also reported as the most frequently isolated human serovar in other countries (Gianfranceschi et al., 2009; Lyytikäinen et al., 2006; Lopez-Valladares et al., 2014). Strains of serotype 1/2a and 1/2c belonged to lineage II, whereas strains of serotype 1/2b and 4b belonged to lineage I. No specific correlation was found between lineage or serotype and clinical presentation.
Antimicrobial susceptibility testing
All 93 L. monocytogenes strains were susceptible to 8 antimicrobial agents (ciprofloxacin, rifampicin, gentamicin, ampicillin, erythromycin, vancomycin, tetracycline, sulfamethoxazole/trimethoprim), and all but 3 (different sequence types) strains were resistant to clindamycin.
PFGE subtyping and cluster analysis
The PFGE fingerprint patterns clustered to form 63 (ApaI) and 70 (AscI) distinguishable pulsotypes at a cut-off value of 95% pattern similarity, which highlights the heterogenicity among the 93 L. monocytogenes strains. Simpson's index of discrimination for ApaI and AscI was 98.7% and 98.8%, respectively. One cluster, formed by the six Swiss 2011 listeriosis outbreak strains (Hächler et al., 2013), and 10 other clusters, formed by two to four strains each, had identical patterns when analyzed with both enzymes. The few other strains, exhibiting identical patterns when analyzed with one enzyme (ApaI), usually yielded different patterns after the second (AscI) enzyme was applied. The high genetic diversity among the strains indicates that L. monocytogenes infections in Switzerland most often occur as single cases.
MLST typing
The 93 isolates were distinguished into 35 STs, 8 of which (ST671, 672, 673, 681, 682, 683, 684, 685) were designated for the first time. Simpson's index of discrimination was 93.9%. ST8 (14 strains, all serotype 1/2a) and ST1 (14 strains, all serotype 4b) were the most prevalent STs identified among these strains. No correlation was found between ST and clinical presentation. Figure 1 represents the diversity of Swiss strains superposed with the diversity described in previous studies (Ragon et al., 2008; Chenal-Francisque et al., 2011; Cantinelli et al., 2013; Chenal-Francisque et al., 2013). The STs found in this study are largely distributed across the clonal diversity of L. monocytogenes, including global collections. These results show that the population of Swiss L. monocytogenes strains is representative of the known diversity of this species, showing that the “everything is everywhere” paradigm of L. monocytogenes clones (Chenal-Francisque et al., 2011; Cantinelli et al., 2013) applies to Switzerland (Fig. 1) and demonstrates the presence of all major L. monocytogenes clones in Switzerland.
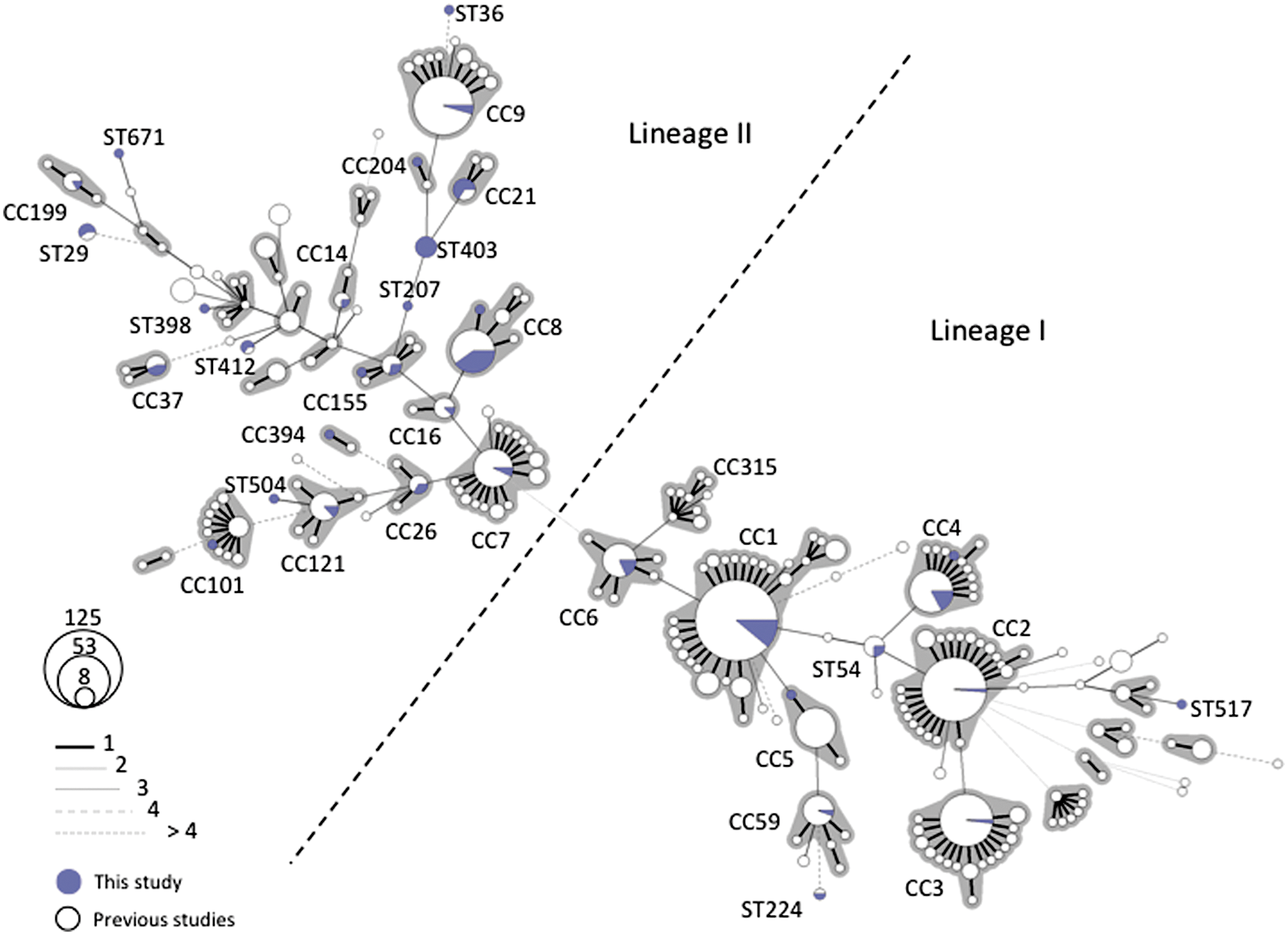
Minimum spanning tree of multilocus sequence typing data for 956 Listeria monocytogenes strains. Each circle represents one sequence type (ST), the size of which is related to the number of strains with this ST. Blue sectors represent the 93 Swiss L. monocytogenes strains from this study, whereas white sectors represent previously published strains (Ragon et al., 2008; Chenal-Francisque et al., 2011; Cantinelli et al., 2013; Chenal-Francisque et al., 2013). Links between circles are represented according to the number of allelic mismatches between STs, as indicated. Gray zones surrounding groups of STs represent clonal complexes (CC).
Internalin A
Internalin A is a cell-wall-anchored protein that is critical to the virulence of L. monocytogenes (Lecuit et al., 2001; Seveau et al., 2007). Several types of virulence-attenuating mutations causing premature stop codons (PMSCs) in the inlA gene have been documented (Jonquieres et al., 1998; Nightingale et al., 2005; Nightingale et al., 2008; Ragon et al., 2008; Cantinelli et al., 2013). Of the 35 examined L. monocytogenes strains, representing each of the STs identified, there were only 3 (8.6%) with inlA gene PMSC mutations. The first strain (N13-1407) belonged to ST199/serotype 1/2a and had a deduced stop codon at amino acid position 9 caused by A deletion at nucleotide position 12 of the inlA gene. The second strain (N12-0367), belonging to ST121/serotype 1/2a, had a deduced stop codon at amino acid position 492 due to a C-to-T mutation at nucleotide position 1474. The third strain (N12-0486), belonging to ST9/serotype 1/2c, had a stop codon at amino acid position 577, precipitated by an A deletion at nucleotide position 1637. Similar PMSCs have been described previously among clinical L. monocytogenes strains isolated from other countries (Felicio et al., 2007; Ragon et al., 2008; Van Stelten et al., 2010; Cantinelli et al., 2013; Kovacevic et al., 2013). All strains with PMSCs belonged to lineage II and were isolated from blood. The low frequency of PMSCs inlA as detected among the sequenced Swiss strains is consistent with previous reports among clinical strains from other countries and shows that isolates from clinical infections most frequently are able to produce full-length InlA (Jacquet et al., 2004; Van Stelten et al., 2010).
Listeriolysin S (LLS)
Listeriolysin S (LLS) is an oxidative stress–inducible hemolysin that contributes to virulence and is present in a subset of lineage I strains (Cotter et al., 2008). PCR screening for the arrangement and presence of llsA, llsG, llsD, llsP genes within the Listeria pathogenicity island 3 showed that LLS-positive strains accounted for 29 (87.8%) of the 33 genetic lineage I strains examined (Table 1). Similar to previous observations (Cotter et al., 2008), this virulence factor was absent in lineage II strains. The positive strains were from serotypes 4b (93.1%) and 1/2b (6.9%), and included representatives from 7 STs. Most of the LLS-positive strains (86.2%) belonged to CC1, CC4, and CC6 (Table 1).
Listeria genomic island (LGI1)
The 50-kb LGI1 consists of genes encoding for putative type II and type IV secretion systems, pilus-like surface structures, a multidrug efflux pump homologue (EmrE), and an alternative sigma factor (Gilmour et al., 2010). Although the exact role of this island has not yet been fully elucidated, it is suggested that it may play a role in bacterial persistence and/or pathogenicity, and it was initially identified in L. monocytogenes serotype 1/2a strains of CC8 responsible for the 2008 Canadian listeriosis outbreak (Gilmour et al., 2010). All of the 58 Swiss serotype 1/2a strains, including those of CC8, were negative for this genomic island using the LGI1 PCR (Table 1). This is worth mentioning as it suggests distinct subpopulations of CC8 strains.
Stress survival islet (SSI-1)
Genes within the stress survival islet (SSI-1) have been suggested to contribute to survival and growth of L. monocytogenes under stress associated with food environments (Ryan et al., 2009). The distribution of this islet was examined among the Swiss listeriosis strains. Its presence was detected in 39 (42%) strains, of which most belonged to serotypes 1/2a (n=33) and 1/2b (n=4) (Table 1). The distribution of this islet among Swiss clinical strains seems similar to previous reports in which this islet was found more prevalent among nonserogroup 4 strains (Jacquet et al., 2004; Kovacevic et al., 2013). For 14 (15.1%) strains, all belonging to serotype 1/2a, there were no SSI-1 PCR amplicons detected, suggesting that the SSI-1 primer binding sites in these strains are incompatible (Table 1). Furthermore, in three serotype 1/2a strains from ST121 and ST504, the amplicon size obtained differed from that expected in the presence (9.7 kb) or absence (1.1 kb) of the SSI-1 islet. These strains gave amplicons of about 2.2 kb in size. Amplicons of similar size were previously reported in some L. monocytogenes strains that belong to serotype 1/2a and ST121 that were found to harbor homologs of L. innocua genes instead of SSI-1 genes in this region (Hein et al., 2011).
In conclusion, the present study delivers insights into the relationships and virulence-associated characteristics of clinical L. monocytogenes strains occurring in Switzerland. No links between serotypes, lineages, or MLST types on the one hand, and clinical origin of the strains on the other hand, were found. The MLST sequence types found in Switzerland are largely distributed across the global clonal diversity of L. monocytogenes. The results further highlight strain differences in the occurrence of several genetic elements, which are linked to bacterial persistence and/or pathogenicity.
Footnotes
Acknowledgments
Herbert Haechler and Grethe Sägesser, National Reference Laboratory, Switzerland, are kindly acknowledged for their technical support and for serotyping of the strains. This work was partly supported by the Swiss Federal Office of Public Health, Division of Communicable Diseases.
Disclosure Statement
No competing financial interests exist.