
Abstract
Shiga toxin–producing Escherichia coli (STEC) is defined by the ability to produce one or more types of Shiga toxins. In an attempt to better understand the mechanisms that underlie pathogenicity among STEC foodborne infection, we compared different STEC serotypes recovered from food sources (O26:H11, O103:H2, and O157:H7) for their interaction with human intestinal epithelial cells using the Caco-2 cell line as an infection model. Bacterial uptake was determined using gentamicin protection assay and results were confirmed by fluorescent microscopy. Our results revealed no significant difference in adherence among tested serotypes. Nonetheless, E. coli O157:H7 exhibited a significant increase of internalization ability and survived significantly better over different time points for up to 24 h. To study cellular invasion mechanisms, multiple inhibitors with known effects on eukaryotic cell structures and processes were used. Inhibition of bacterial and host cell protein synthesis significantly diminished entry in all tested serotypes, suggesting that invasion is an active process that requires both bacterial and eukaryotic protein syntheses. Cytochalasin D (an actin microfilament inhibitor) and staurosporine (inhibitor of several protein kinases) significantly decreased internalization of all tested serotypes. Our results suggested that invasion by STEC varies between different serotypes, might correlate with clinical outcome, and is receptor mediated. This process is dependent on microfilament-dependent pathway and phosphorylation of cell proteins but not on formation of microtubules.
Introduction
S
The genes and mechanisms that enable STEC to colonize the human intestine are incompletely understood (Cordeiro et al., 2013). Adherence of enteric pathogens to epithelial surfaces is often a prerequisite for the subsequent delivery of bacterial enterotoxins and mucosal invasion and is frequently a critical step in bacterial replication and colonization of the gastrointestinal tract (Sherman et al., 1988). This could explain the high rate of progression to HUS caused by STEC infection (Bielaszewska et al., 2011). Furthermore, other studies moved from the role played by bacterial adhesins to the investigation of STEC invasiveness in colonization and infection (Sheng et al., 2011).
In this study, we aimed to study the mechanisms that might underlie variation in ability of different STEC serotypes to cause human infection and to determine invasion-associated properties of these serotypes to human intestinal epithelial cells. This was achieved by comparing intestinal adherence, invasion, and survival properties of three different STEC serotypes (O26:H11, O103:H2, and O157:H7) recovered from different food sources using the enterocyte-like Caco-2 cell line and by assaying the invasion using different selective agents with known effects on bacteria and eukaryotic cells.
Materials and Methods
Bacterial strains and culture conditions
Seven STEC strains belonging to three different serotypes were used in this study (Table 1). Isolates were recovered from raw-milk cheese (Kareish and Damietta cheese) and retail beef products during previous separate studies (Elhadidy and Mohammed, 2013; Sallam et al., 2013). STEC strains were identified using API 20E biochemical test strips (BioMérieux, Marcy-l'Etoile, France), serotyping of somatic lipopolysaccharide (O) and flagellar (H) antigens, and PCR amplification of stx1, stx2, and eae genes (Elhadidy and Mohammed, 2013). For invasion experiments, Salmonella Typhimurium ATCC 14028 and E. coli K12 (DH5α) were used as positive and negative controls, respectively (Cordeiro et al., 2013). All isolates were stored at −20°C in 20% glycerol–brain heart infusion broth (BHI) for further testing. All bacterial subcultures were performed in BHI and agar plates (Oxoid). Bacterial concentrations were determined by densitometry (OD600) reading and plate counting assay.
+, eae positive.
stx1, Shiga toxin 1; stx2, Shiga toxin 2.
Tissue culture
Caco-2 human colon adenocarcinoma cell line (HTB37, ATCC, USA) was used in this study due to its high similarity to human enterocytes with respect to their structure, brush border enzymes, and time courses of differentiation after incubation for 12 days (Szymanski et al., 1995). The cell line was grown and maintained as previously described (Cordeiro et al., 2013). Cell viability was detected using trypan blue staining. Prior to passage, cells were washed with calcium-free Dulbecco's phosphate-buffered saline (D-PBS; Sigma-Aldrich, MO). Cells were released from plastic by rinsing with trypsin solution (0.5% trypsin in D-PBS) for approximately 30 s. Trypsin was aspirated, and flasks were incubated at 37°C until cells were detached. Trypsinization was stopped by addition of growth medium plus 10% (vol/vol) fetal bovine serum.
Assessment of bacterial adherence to Caco-2 cells
Quantitative adherence experiments were carried out using viable count assay to quantify the total cell-associated bacteria (intracellular bacteria plus surface-adherent bacteria). Caco-2 cells were plated (approximately 5×104 cells/well) in 24-well tissue culture plates (Corning Inc., Corning, NY) and maintained for 12 d for differentiation. Prior to infection, the growth media were removed and the wells were washed 3 times with D-PBS to remove antibiotic residues and monolayers were covered with 500 μL of fresh Dulbecco's Modified Eagle medium (DMEM). All tested strains were grown from frozen stocks onto BHI agar plates and incubated at 37°C for 18 h. Suspension from colonies were made in 1-mL D-PBS at a concentration adjusted to 106 colony-forming units (CFU)/mL. For each tested strain, 500 μL was suspended in 500 μL of fresh DMEM and used to infect confluent monolayers. The monolayers were then incubated at 37°C for 6 h with medium change after 3-h incubation to minimize extracellular bacterial growth. After incubation, all wells were washed 3 times with D-PBS to remove nonadherent bacteria and 1 mL of 1% (vol/vol) Triton X-100 in D-PBS was added for eukaryotic cell lyses and further incubated at 37°C for 30 min. Lysates were serially diluted and 10 μL of dilutions was plated into BHI agar plates. Adhesion was expressed as the total number of CFU/well. Each strain was assayed in triplicate wells, and the assay was repeated three times.
Assessment of bacterial invasion
Bacterial uptake was performed using gentamicin protection assay as previously described (Yamamoto et al., 2009) with some modifications. After the 6-h infection period mentioned above, the monolayers were washed 3 times with D-PBS and 250 μL fresh medium DMEM plus gentamicin sulfate (100 μg/mL) were added to all wells and incubated for 1 h to eliminate viable extracellular bacteria. All strains were tested to be susceptible to 100 μg/mL of gentamicin sulfate prior to the invasion experiments. Control wells were incubated with medium without antibiotics. After incubation, 10 μL was plated on BHI agar plates to check killing of extracellular bacteria. Wells were washed 4 times with D-PBS to remove antibiotic residues and 1 mL of 1% (vol/vol) Triton X-100 in D-PBS was added to wells, incubated for 30 min, and then serially diluted and plated on BHI agar plates. The number of intracellular bacteria was expressed as the total number of CFU/well.
Assessment of bacterial intracellular survival
To compare the survival capabilities of tested strains within Caco-2 cells, infected monolayers were further incubated for different time points up to 24 h under gentamicin protection treatment. After the infection of cells for 6 h, cells were washed once in D-PBS and 1 mL of fresh infection medium with gentamicin sulfate (40 μg/mL) were added to the monolayer for 4, 8, 12, and 24 h. At each time point, cells were washed, lysed, and the cell lysates were diluted and plated on BHI agar plates.
Effect of bacterial and eukaryotic cell inhibitors on bacterial invasion
To test the contribution of both bacterial and host cell proteins in the invasion process, quantitative invasion assays were performed in the presence of 100 μg/mL chloramphenicol (bacterial protein synthesis inhibitor) or 20 μg/mL cycloheximide (eukaryotic protein synthesis inhibitor) during the bacterial infection (6 h) and gentamicin treatment periods (1 h). To test the participation of host cell cytoskeleton components in the invasion of tested serotypes, Caco-2 cells were incubated 30 min prior to bacterial inoculation with 2 mg/mL, 5 mg/mL, and 0.5 mg/mL of cytochalasin-D (actin microfilament polymerization inhibitor), colchicine
Effect of inhibitors on bacterial and eukaryotic cell viabilities
The viabilities of bacteria and Caco-2 cells were determined following incubation in media containing the mentioned concentration of each inhibitor that gives maximal inhibitory activity without affecting cell viability for the abovementioned incubation time. Bacterial viability was also tested following incubation with 1% (vol/vol) Triton X-100 for 30 min. Bacterial viability was assessed using viable counts on agar medium; viability of cells was determined by counting cells in a hemocytometer after staining with trypan blue dye.
Microscopy
Double immunofluorescence was performed to confirm invasion and invasion inhibition results. Round coverslips (12-mm diameter) were placed in the wells of a 24-well tissue culture plate, seeded with 5×104 Caco-2 cells/well, and incubated with tested strains at 5×105 CFU/mL for required time of invasion/survival experiments mentioned above. After infection, coverslips were removed, and cells were fixed with formaldehyde (4% in PBS) for 15 min and washed 3 times with PBS, permeabilized for 2 min with 0.1% (vol/vol) Triton X-100 in D-PBS, blocked with 5% bovine serum albumin (BSA) in D-PBS for 30 min to block nonspecific binding sites and incubated with O26:H11, O103:H2, and O157:H7 rabbit antisera diluted 1:50 in 1% (vol/vol) BSA in D-PBS (PBS-BSA) for 1 h at 37°C. Caco-2 cells were washed again three times with PBS and Alexa 488-conjugated goat anti-rabbit IgG (Molecular Probes, Carlsbad, CA) diluted 1:200 in 1% (vol/vol). BSA-PBS was added and incubated at 37°C for 1 h. Cells were counterstained with 4'6-diamidino-2-phenylindole (Molecular Probes) for 3 min. Coverslips were removed and washed three times with PBS. Slides were examined by fluorescence microscopy, and the number of bacteria per field was counted for randomly selected fields in each well for at least 20 fields.
Statistics
All results were analyzed with Microsoft Excel (Microsoft Corp., Redmond WA), two-tailed Student t-tests, and a probability of <0.05 was considered significant.
Results and Discussion
In this study, a quantitative in vitro cell culture infection model was used to study the mechanism of interaction of different STEC serotypes with intestinal epithelial cells. The serotypes O26:H11, O103:H2 were used for comparison together with O157:H7 because these serotypes are considered the most important non-O157 STEC serotypes associated with increasing frequency in patients with bloody diarrhea and HUS (Jelacic et al., 2003; Brooks et al., 2005; Mellmann et al., 2005). First, we studied these organisms by investigating their adhesion and invasion properties to human Caco-2 cells originating from the colon. This cell line is commonly used as a model system simulating STEC interaction with intestinal epithelial cells in vivo (McKee and O'Brien, 1995; Cordeiro et al., 2013). To exclude the possibility that the difference in our quantitative experiments is due to variation in growth phase or cell densities among strains, we confirmed that the growth rates of each bacterial culture were similar and the CFU/mL of the starting inocula varied by <10% (data not shown). In addition, our quantitative experiments showed no significant difference between different strains within the same serotype (p>0.5)
We first analyzed the ability of the tested STEC serotypes to adhere to Caco-2 cell line using viable plate counts, and results were expressed as CFU/well. Mean level of adherence of O26:H11, O103:H2, and O157:H7 to Caco-2 cells was 1.83×105, 1.72×105, and 2.10×105 CFU/well, respectively (Fig. 1). Our results showed no significant difference in the mean level of adherence to intestinal epithelial cells between different tested serotypes (p>0.2) (Fig. 1). These results suggested that all tested serotypes behave the same in their ability to initiate the steps required for pathogenesis and establishment of infection.
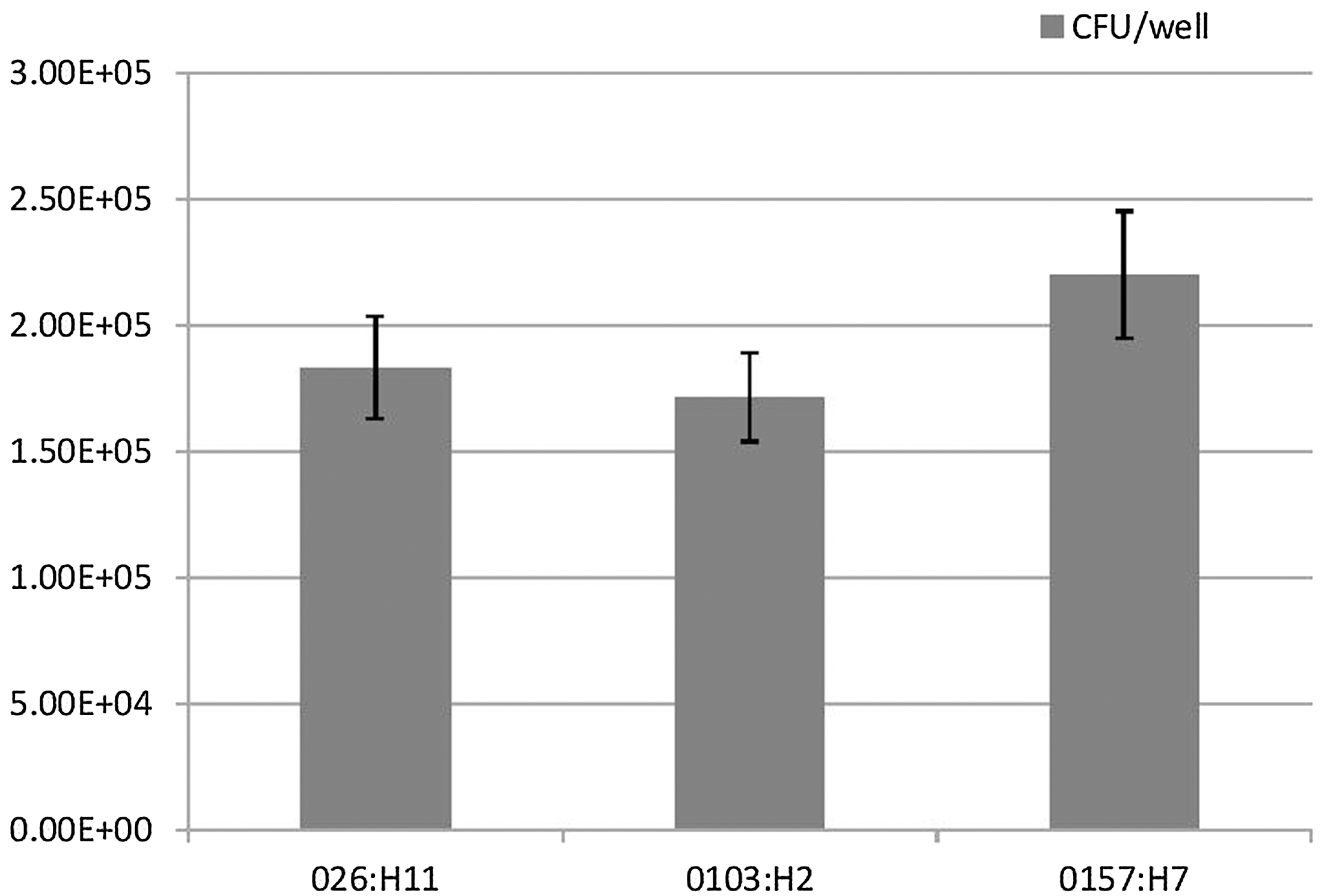
Association of Shiga toxin–producing Escherichia coli O26:H11, O103:H2, and O157:H7 strains with Caco-2 cells. Cell attachment was determined by viable plate counts. Adhesion results were expressed as the total number of colony-forming units (CFU)/well. There was no significant difference in the mean level of adherence to Caco-2 cells between different tested serotypes (p>0.2). Results represent the means and standard deviations of three independent experiments performed in triplicate.
Quantitative assessment of invasive bacteria (bacteria that are able to induce their own phagocytosis into cells that are normally nonphagocytic) was performed by gentamicin protection assay and results were confirmed by immunofluorescence. E. coli O157:H7 exhibited a significant increase in the uptake efficiency compared to other non-O157:H7 serotypes tested (O26:H11 and O103:H2 serotypes) (p<0.0001). The mean value of invasion by E. coli O157:H7 strains was 2.5-fold higher than the other non-O157:H7 strains tested (Fig. 2). Together with the adherence data, these results suggested that the adhesive ability does not translate directly into invasive efficiency as all other tested serotypes showed same level of adhesion. No significant difference in invasion abilities was observed between O26:H11 and O103:H2 serotypes (p>0.2). Invasion of Salmonella Typhimurium–positive control was two times more than mean values of E. coli O157:H7 strains. No invasion was detected for E. coli DH5α (negative control). Overall, these findings, coupled with previous studies reporting that E. coli O157:H7 appeared to be more clinically severe than non-O157 STEC infections (Wang et al., 2013), might point to differences in the expression of specialized ligands mediating invasion by E. coli O157:H7 that mediates human infection. Further work using gene expression and microarray analysis together with in vivo experiments is crucial to prove this postulation. Another explanation for the enhanced invasion ability by E. coli O157:H7 is the polysaccharide side chain that defines this serogroup. This postulation is supported by a previous study that suggested that the O antigen in E. coli O157:H7 contributes to efficient bovine colonization (Sheng et al., 2008). Overall, it remains to be elucidated whether E. coli O157:H7 strains bear additional and/or specific virulence properties or different invasive mechanisms that are not present in other tested serotypes or that vary from one serotype to another that could allow them to better invade human enterocytes.
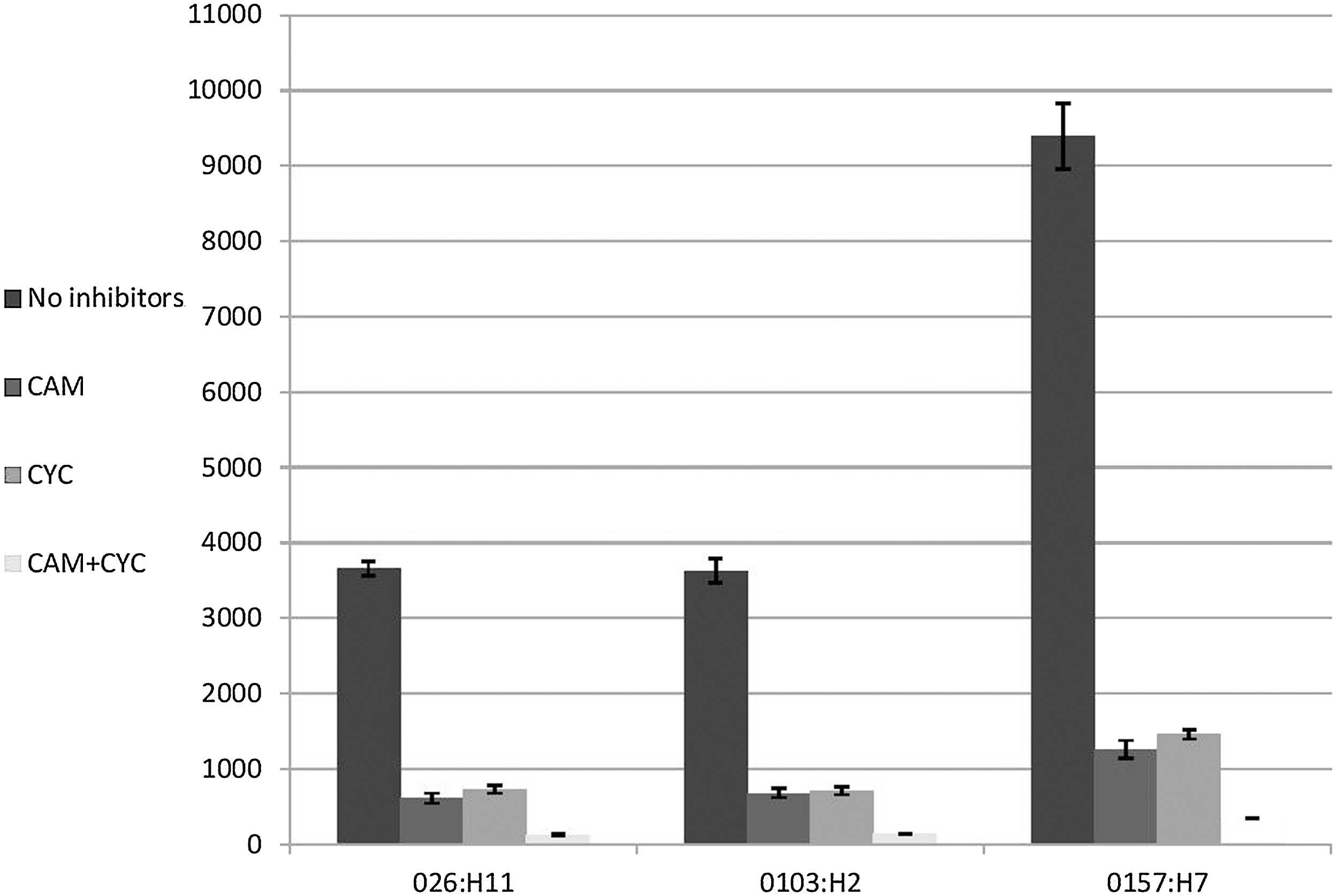
Invasion of Shiga toxin–producing Escherichia coli O26:H11, O103:H2, and O157:H7 strains to Caco-2 cells using the gentamicin protection assay. Invasion results were expressed as the total number of colony-forming units/well. E. coli O157:H7 exhibit a more significant increase in the efficiency of invasion than the other non-O157:H7 serotypes tested (O26:H11, and O103:H2 serotypes) (p<0.0001). Invasion of Caco-2 cells was significantly decreased in all tested serotypes following inhibition of both bacterial and eukaryotic proteins by chloramphenicol (CAM) and cyclohexamide (CYC), respectively (p<0.0001). Results represent the means and standard deviations of three independent experiments performed in triplicate.
Invasion of intestinal epithelial cells can provide bacteria with a survival advantage for enteric pathogens, allowing them to better resist detection and clearance by both innate and adaptive immune defense mechanisms and antibiotics, enabling these pathogens to cause persistent and chronic infections (Finlay and Falkow, 1997). To further examine whether such invaded bacteria were able to survive inside intestinal epithelial cells, we examined intracellular survival at multiple time points after infection and lysis of eukaryotic cells by Triton-X 100. Our results revealed that E. coli O157:H7 strains had a higher survival rate inside the Caco-2 cell line than non- O157:H7 strains by 3.1–3.6 times over a 24-h incubation period (p<0.001) (Fig. 3). No significant difference in survival abilities was observed between O26:H11 and O103:H2 serotypes (p>0.05) (Fig. 3).
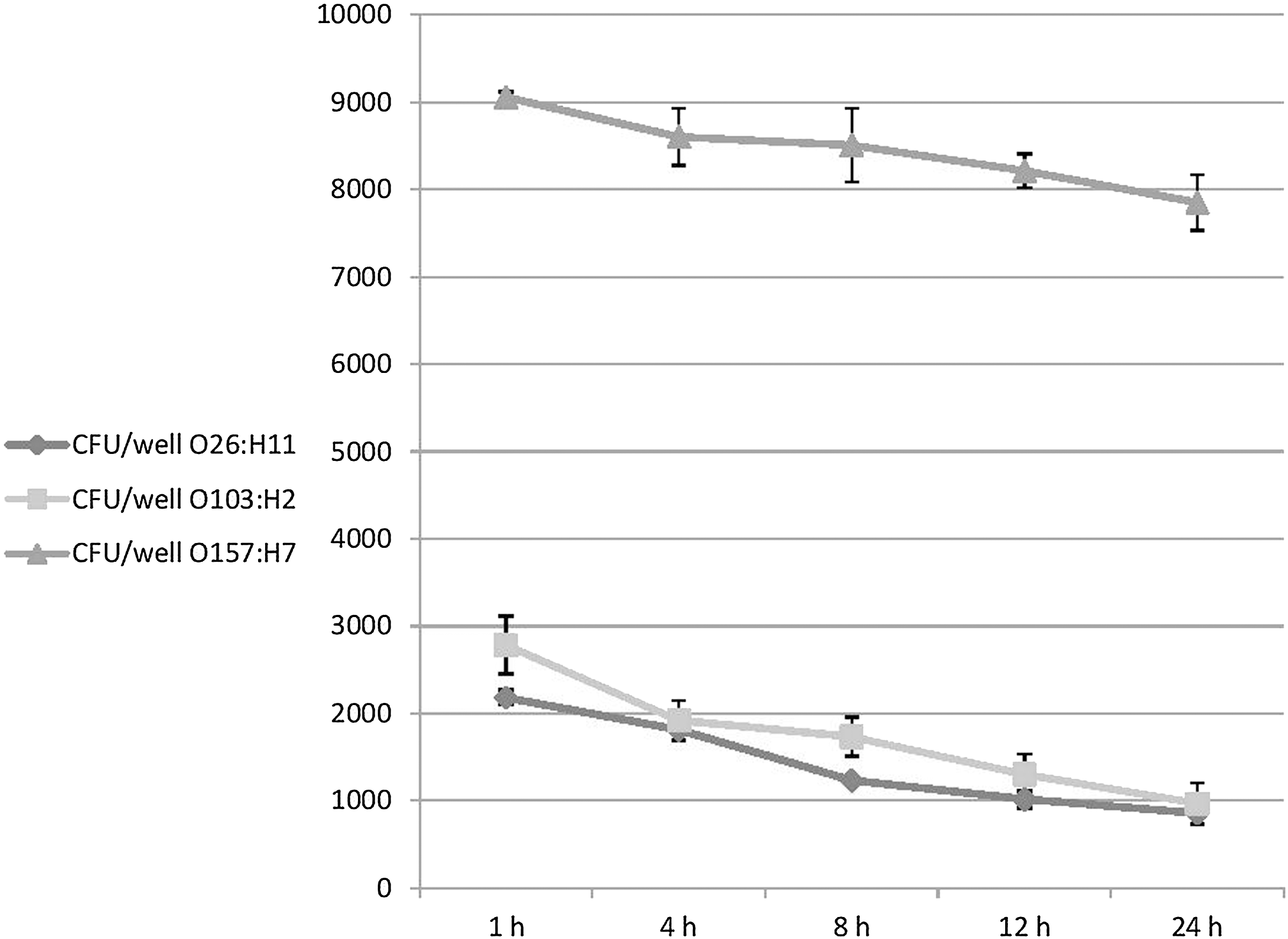
Survival of Shiga toxin–producing Escherichia coli O26:H11, O103:H2, and O157:H7 strains inside Caco-2 cells using a gentamicin protection assay at different incubation time points up to 24 h (1 h, 4 h, 8 h, 12 h, and 24 h). Survival results were expressed as the total number of colony-forming units/well. E. coli O157:H7 strains are better able to survive inside the Caco-2 cell line than non- O157:H7 strains over a 24-h time period (p<0.001). Results represent the means and standard deviations of three independent experiments performed in triplicate.
To determine whether bacterial and host cell protein synthesis are required for invasion, chloramphenicol and cyclohexamide, which are inhibitors specific for prokaryotic and eukaryotic protein synthesis, respectively (Finlay, 1990), were added to the bacterial inoculum immediately before Caco-2 cells were infected. Chloramphenicol and cycloheximide were used to inhibit bacterial and eukaryotic protein biosynthesis by binding to the ribosomal subunits. Chloramphenicol had no significant effect on bacterial viability for the required incubation time (data not shown). The minimum inhibitory concentrations (MICs) of chloramphenicol for tested strains were ≥30 μg/mL (data not shown). However, the working concentration used in this study (100 μg/mL) did not kill tested strains for the required incubation period (data not shown). Consistent with this finding and in an earlier invasion study, Kim and Loessner, 2008, reported that tested Enterobacter sakazakii strains were not killed by chloramphenicol at 100 μg/mL following incubation for 3 h while tested MICs for Enterobacter sakazakii were 4–8 μg/mL. Inhibition of both bacterial and eukaryotic proteins significantly inhibited invasion of Caco-2 cells in all tested serotypes by 5.3–7.5- and 5–6.4-fold, respectively (p<0.0001) (Fig. 2). These results indicated that invasion of tested serotypes to enterocytes is an active process that requires a pool of bacterial and host cell proteins. A similar active invasion process was reported in other enteric pathogens including Shigella spp. (Hale et al., 1979) and Salmonella spp. (Finlay and Falkow, 1997) but not Yersinia enterocolitica (Finlay and Falkow, 1997).
In order to further identify the host cell structures and processes that might be involved in Caco-2 cells invasion by STEC, Caco-2 cells were treated with reagents affecting the cytoskeleton such as cytochalasin D (to disrupt actin microfilament formation by capping the growing ends of actin filaments, causing depolymerization of actin filaments), staurosporine (inhibitor of several protein kinases needed for transducing extracellular signals), or colchicine (inhibitor of microtubule function by interfering with tubulin polymerization) prior to infection. These inhibitors were added to Caco-2 cells for 30 min prior to addition of bacteria. None of these inhibitors affected bacterial or cell viability or adhesion to Caco-2 cells for the required incubation time (data not shown). Infection of eukaryotic host cells pretreated with the actin polymerization inhibitor cytochalasin D significantly decreased the mean levels of invasion of STEC serotypes O26:H11, O103:H2, and O157:H7 by 3.8-, 4.5-, and 4.4-fold, respectively (p<0.0001) (Fig. 4). Similarly, pretreatment of eukaryotic host cells protein kinases inhibitor staurosporine significantly decreased the mean levels of invasion of STEC serotypes O26:H11, O103:H2, and O157:H7 by 2.2-, 2.8-, and 5.7-fold, respectively (p<0.0001) (Fig. 4). On the other hand, Caco-2 pretreatment with microtubule inhibitor colchicine resulted in nonsignificant inhibition in invasion process by all tested serotypes (p>0.2) (Fig. 4). These results suggested that entry of all tested STEC serotypes to Caco-2 cells was dependent on eukaryotic microfilament assembly and phosphorylation of cell proteins by protein kinases but not microtubule formation. These results are consistent with previous studies reporting involvement of host cell cytoskeletal rearrangement and phosphorylation-mediated signal transduction in cell invasion by STEC (Cordeiro et al., 2013). However, whether these cytoskeleton structures are also needed in other cell types remains to be elucidated in future studies.
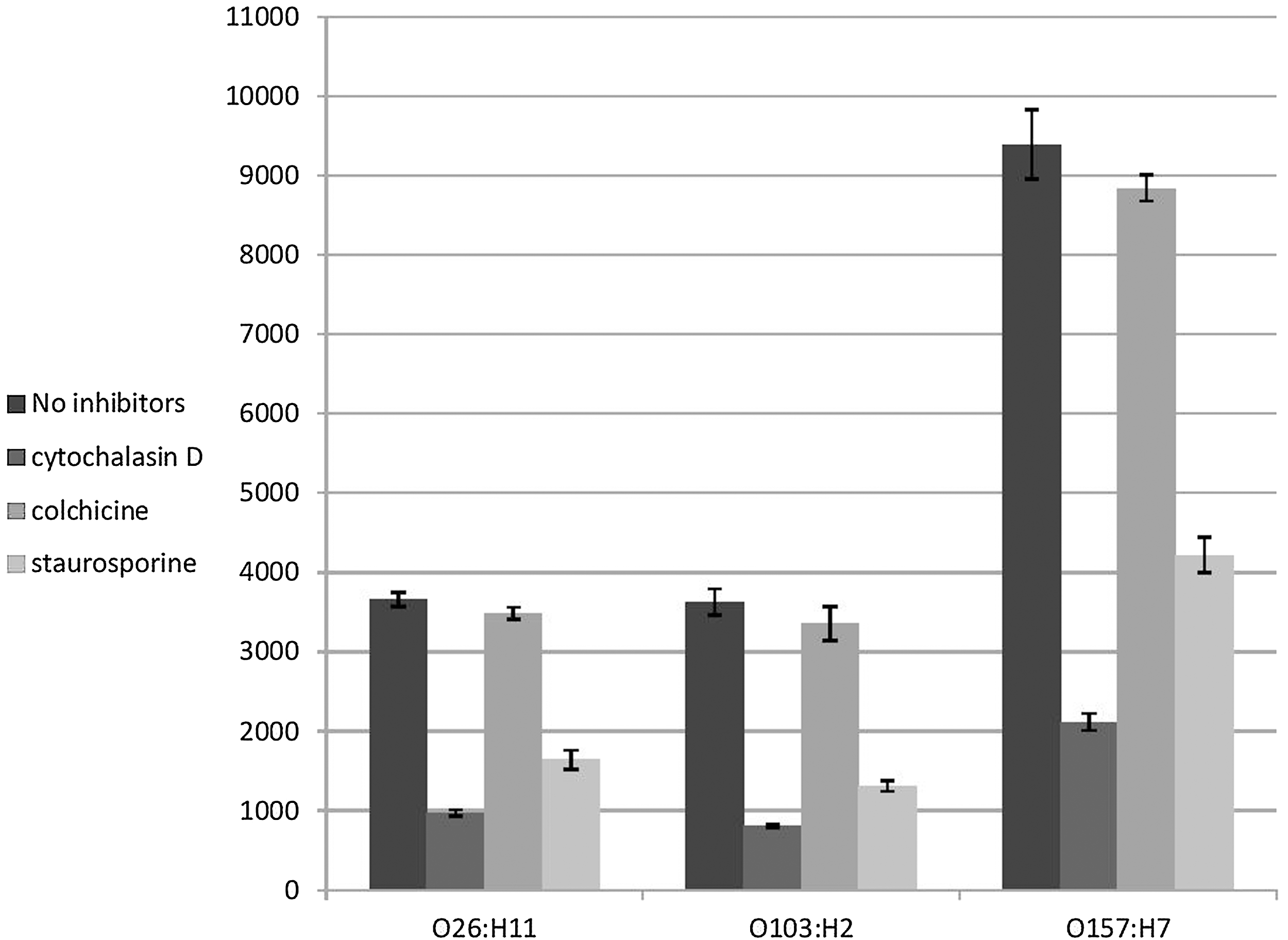
Invasion of Shiga toxin–producing Escherichia coli O26:H11, O103:H2, and O157:H7 strains to Caco-2 cells following inhibition of eukaryotic cells by cytochalasin D (an actin microfilament inhibitor), colchicine (eukaryotic microtubule function inhibitor), and staurosporine (inhibitor of several protein kinases). Treatment of Caco-2 cells with cytochalasin D and staurosporine significantly decreased invasion of all tested serotypes (p<0.0001). Colchicine had no effect on the invasion process by tested serotypes (p>0.2). Results represent the means and standard deviations of three independent experiments performed in triplicate.
Conclusions
We succeeded in highlighting some important requirements of invasion of some STEC serotypes to human intestinal epithelial cells using Caco-2 cells, which provides a step that should assist in determining the molecular requirements for STEC pathogenesis. Invasion in all tested serotypes appears to be receptor mediated and requires actin filaments but not microtubules formation. The significant increase in invasion and survival ability of E. coli O157:H7 strains inside Caco-2 cells suggest one of the possible reasons for increased disease severity of this serotype compared to other serotypes. Future studies should be directed to identify bacterial and host cell invasion-associated molecules and to demonstrate entry and colonization mechanisms into other cell types to assist in building a solid foundation of STEC pathogenesis mechanisms.
Footnotes
Acknowledgment
The authors would like to thank Professor Khaled Sallam and Dr. Asmaa Ahdy, Department of Food Hygiene and Control, Mansoura University, for providing E. coli O157:H7 strains and for helpful discussions.
This work was presented in part at ESCMID Conference of Escherichia coli: An Old Friend With New Tidings, held in Barcelona November 20–22, 2013 and at the 18th Conference on Food Microbiology, Brussels, Belgium September 12–13, 2013.
Disclosure Statement
No competing financial interests exist.