
Abstract
Salmonella enterica subspecies enterica serotype Newport is one of the common serotypes causing foodborne salmonellosis outbreaks in the United States. Salmonella Newport consists of three lineages exhibiting extensive genetic diversity. Due to the importance of Salmonella pathogenicity islands 5 and 6 (SPI-5 and SPI-6) in virulence of pathogenic Salmonella, the genetic diversity of these two SPIs may relate to different potentials of Salmonella Newport pathogenicity. Most Salmonella Newport strains from North America belong to Salmonella Newport lineages II and III. A total 28 Salmonella Newport strains of lineages II and III from diverse sources and geographic locations were analyzed, and 11 additional Salmonella genomes were used as outgroup in phylogenetic analyses. SPI-5 was identified in all Salmonella Newport strains and 146 single nucleotide polymorphisms (SNPs) were detected. Thirty-nine lineage-defining SNPs were identified, including 18 nonsynonymous SNPs. Two 40-kb genomic islands (SPI5-GI1 and SPI5-GI2) encoding bacteriophage genes were found between tRNA-ser and pipA. SPI5-GI1 was only present in Salmonella Newport multidrug-resistant strains of lineage II. SPI-6 was found in all strains but three Asian strains in Salmonella Newport lineage II, whereas the three Asian strains carried genomic island SPI6-GI1 at the same locus as SPI-6 in other Salmonella. SPI-6 exhibited 937 SNPs, and phylogenetic analysis demonstrated that clustering of Salmonella Newport isolates was a reflection of their geographic origins. The sequence diversity within SPI-5 and SPI-6 suggests possible recombination events and different virulence potentials of Salmonella Newport. The SNPs could be used as biomarkers during epidemiological investigations.
Introduction
N
Salmonella Newport consists of three lineages with extensive genetic diversity (Sangal et al., 2010). Most Salmonella Newport strains from Europe belong to lineage I, whereas most North American strains belong to lineages II and III (Sangal et al., 2010). Whole genome sequence analysis of 28 Salmonella Newport strains from diverse sources and locations grouped the strains into lineages II and III with clustering explained by geographic origins (Cao et al., 2013). The Asian strains were clustered separately from American strains. To arrive at a comprehensive evolutionary picture of Salmonella Newport, it would be necessary to include all three lineages; however, no Salmonella Newport lineage I strain has been sequenced to date.
Genomic island (GI) is a gene cluster that has been acquired via horizontal gene transfer (Langille et al., 2010). Pathogenicity islands are gene clusters encoding virulence determinants that are usually absent in nonpathogenic strains of the same or closely related species (Sabbagh et al., 2010). A total of 22 Salmonella pathogenicity islands (SPIs) have been identified to date (Sabbagh et al., 2010). Salmonella Newport genomes contained SPI-1 through SPI-4 sequences (unpublished data) and showed extensive diversities at the region around mutS downstream of SPI-1 (Cao et al., 2013).
SPI-5 was first identified in the Salmonella Dublin genome between tRNA-serT and copR and was found to consist of five genes (pipA, pipB, pipC, sopB, and pipD) (Wood et al., 1998). These five genes displayed high similarity with genes from bacteriophages Gifsy-1 and Gifsy-2 (Figueroa-Bossi et al., 2001). SPI-5 plays a vital role in pathogenicity and encodes effectors of SPI-1 and SPI-2 (Sabbagh et al., 2010). For example, sopB encodes a translocated effector protein of type III secretion systems (T3SS) in SPI-1 under control of hilA, whereas pipB encodes a translocated effector of T3SS in SPI-2 under control of ssrAB (Knodler et al., 2002; Hensel, 2004). SPI-5 contributes to the colonization of the spleen in chickens (Rychlik et al., 2009). Mutations in SPI-5 genes significantly reduced the enteropathogenicity of Salmonella (Wood et al., 1998).
SPI-6 is located between tRNA-aspV and sinR at centisome 7 in Salmonella encoding a type six secretion system (T6SS) and a Salmonella atypical fimbriae (saf) cluster (Sabbagh et al., 2010). SPI-6 has different gene contents in various serotypes. For example, it is a 47-kb island in Salmonella Typhimurium (Folkesson et al., 1999) and a 59-kb island in Salmonella Typhi (Parkhill et al., 2001). T6SS is widespread in bacteria (Schwarz et al., 2010), and its gene products performs diverse functions (Blondel et al., 2009; Jani and Cotter, 2010), one of which is to mediate antagonistic interactions between bacteria (Hood et al., 2010). Folkesson et al. (Folkesson et al., 2002) reported that the deletion of SPI-6 reduced the invasion activity of Salmonella Typhimurium into Hep2 cells. The saf genes are located downstream of T6SS in SPI-6 and are present in most clinical isolates of Salmonella (Folkesson et al., 1999; Humphries et al., 2003). However, the saf operon encoding nonfimbrial adhesion elements does not contribute to virulence in mice (Folkesson et al., 1999).
The objectives of the current study were to investigate the different virulence potential via identifying genetic diversity in SPI-5 and SPI-6 of Salmonella Newport lineages II and III and to identify markers in SPI-5 and SPI-6 for Salmonella Newport subtyping.
Materials and Methods
Genomes
Twenty-eight Salmonella Newport genomes from diverse sources and locations from our previous work (Table 1) and 11 outgroup genomes were analyzed in the current study (Cao et al., 2013; Lienau et al., 2013; Timme et al., 2013), including Salmonella Tennessee CDC07_0191 (ACBF00000000), Salmonella Kentucky CVM29188 (ABAK00000000), Salmonella Kentucky CDC191 (ABEI00000000), Salmonella Gallinarum 287/91 (AM933173.1), Salmonella Dublin CT02021853 (CP001144.1), Salmonella Hadar RI_05P066 (ABFG00000000), Salmonella Typhimurium LT2 (NC_003197.1), Salmonella Typhimurium SL1344 (NC_016810.1), Salmonella Typhimurium D23580 (NC_016854.1), Salmonella Typhimurium 14028S (CP001363.1) and Salmonella 4,[5],12:i:- SL474 (ABAO00000000).
These genomes were selected from our published study.
Phylogenetic analysis
A whole genome parsimony tree was reconstructed based on 131,855 informative single nucleotide polymorphisms (SNPs) with the Tree analysis using New Technology (TNT) program (Goloboff et al., 2008). The phylogenetic analysis found a minimum tree length with 20 reiterations using Section Search, Ratchet, Drift, and Tree fusing methods, and it calculated 100,000 bootstrapping replicates. Multiple sequence alignment using MULCLE with default parameter (Edgar, 2004) in SEAVIEW (Galtier et al., 1996) identified 146 SNPs in SPI-5, 937 SNPs in SPI-6 (excluding saf genes), and 355 SNPs in saf genes. Parsimony trees of SPI-5, SPI-6, and saf genes were reconstructed using TNT and the same parameters as above. Certain strains were not included in analyses of SPI-5, SPI-6, or the saf genes because of the poor data quality of the draft genomes, such as canine_AZ_2003, bison_TN_2004, and equine_TN_2004_1.
Genetic characterizations of SPI-5 genomic islands 1 and 2 (SPI5-GI1 and SPI5-GI2), and SPI-6 genomic island 1 (SPI6-GI1)
Genetic organizations of SPI5-GIs and SPI6-GI1 were displayed using Mauve (Darling et al., 2004). The best match of genes in SPI5-GI1, SPI5-GI2, and SPI6-GI1 was determined using blastp (Altschul et al., 1990), followed by verification using tblastn (Altschul et al., 1990).
Distance matrix
MEGA 6.05 (Tamura et al., 2011) was used to calculate evolutionary distances (number of differences) over sequence pairs with 10,000 bootstrap iterations for SPI-5, SPI-6, and saf genes.
Results
Phylogenetic tree based on whole genome data
A whole genome phylogenetic tree was constructed using more than 131,855 SNPs (Fig. 1). To better display the evolutionary relationship between Salmonella Newport strains, we selected 11 genomes as outgroups. There were six equally most parsimonious trees with the same branch order at the subgroup level, meaning that the Salmonella Newport strains in each subgroup were the same in all the resulting trees. Salmonella Newport strains were divided into lineages II and III. Lineage II was further grouped into subgroups IIA, IIB, and IIC. All multidrug-resistant (MDR) strains were in node M of subgroup IIC (Cao et al., 2013).
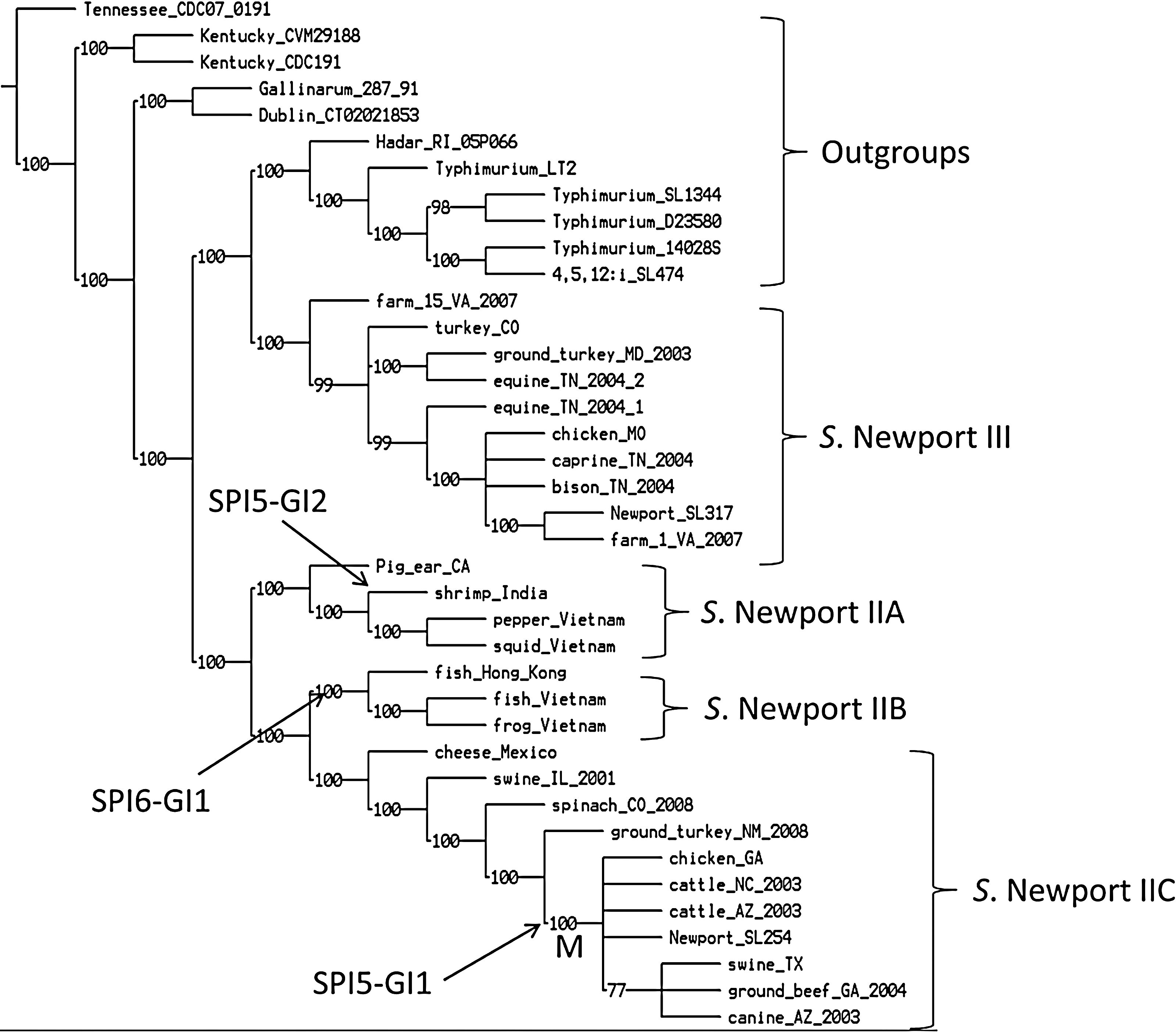
Whole-genome parsimony tree of Salmonella Newport and 11 outgroup genomes. Salmonella Newport strains showed phylogenies identical to those of a previous study (Cao et al., 2013). There are six equally most parsimonious trees identified with a length of 209,114 single nucleotide polymorphisms, consistency index of 0.616, and retention index of 0.888. Two gene clusters, SPI5-GI1 and SPI5-GI2, encoding bacteriophage genes are displayed. The rest of the genomes do not contain SPI5-1 or SPI5-2.
Genetic diversity of Salmonella pathogenicity island 5
SPI-5 was present in 28 Salmonella Newport and 11 outgroup genomes. SPI-5 variations included insertions and SNPs. Two genomic islands encoding prophage genes were found between tRNA-ser and pipA in certain genomes and designated as SPI-5 genomic islands 1 and 2 (SPI5-GI1 and SPI5-GI2) (Supplementary Fig. S1; Supplementary Data are available online at
Some genomes did not contain the entire SPI5-GI1 and SPI5-GI2 between tRNA-ser and pipA; however, partial sequences of SPI5-GIs were identified. For example, a gene cluster in SPI5-GI1 (SNSL254_A1155 to SNSL254_A1177, 5' to 3') was present in Salmonella Typhi CT18, Salmonella Paratyphi B SPB7, Salmonella Paratyphi C RKS4594, and S. Choleraesuis SC-B67. Similarly, part of the SPI5-GI2 sequence (SEEN443_12678 to SEEN443_12753, 5' to 3') was identified in Salmonella Weltevreden HI_N05-537, Salmonella Newport SNSL317, Salmonella Typhimurium DT104, and Salmonella Saintpaul SARA29. The blast matches indicated that 74% and 52% of the genes in SPI5-GI1 and SPI5-GI2, respectively, encoded hypothetical or bacteriophage proteins. Based on current annotation, no gene relating to virulence or antimicrobial resistance was present. Both SPI5-GIs contained genes encoding a methylase (Supplementary Tables S1 and S2).
The five genes in SPI-5 possessed 146 SNPs (Supplementary Table S3). The phylogenetic tree of SPI-5 showed that Salmonella Newport lineages II and III were separated by outgroup genomes (Fig. 2). TNT program identified 227 equally most parsimonious trees with the same taxa at lineage level, meaning that Salmonella Newport isolates in each lineage were clustered together and separated by outgroup in the resulting trees. SNPs in SPI-5 could not distinguish Salmonella Newport at the subgroup level in lineage II. Pairwise distance matrix showed SNPs differences between Salmonella Newport and other serotypes (Table 2). The average differences between lineages II and III were 40 SNPs but only 18 SNPs between Salmonella Typhimurium and lineage II.
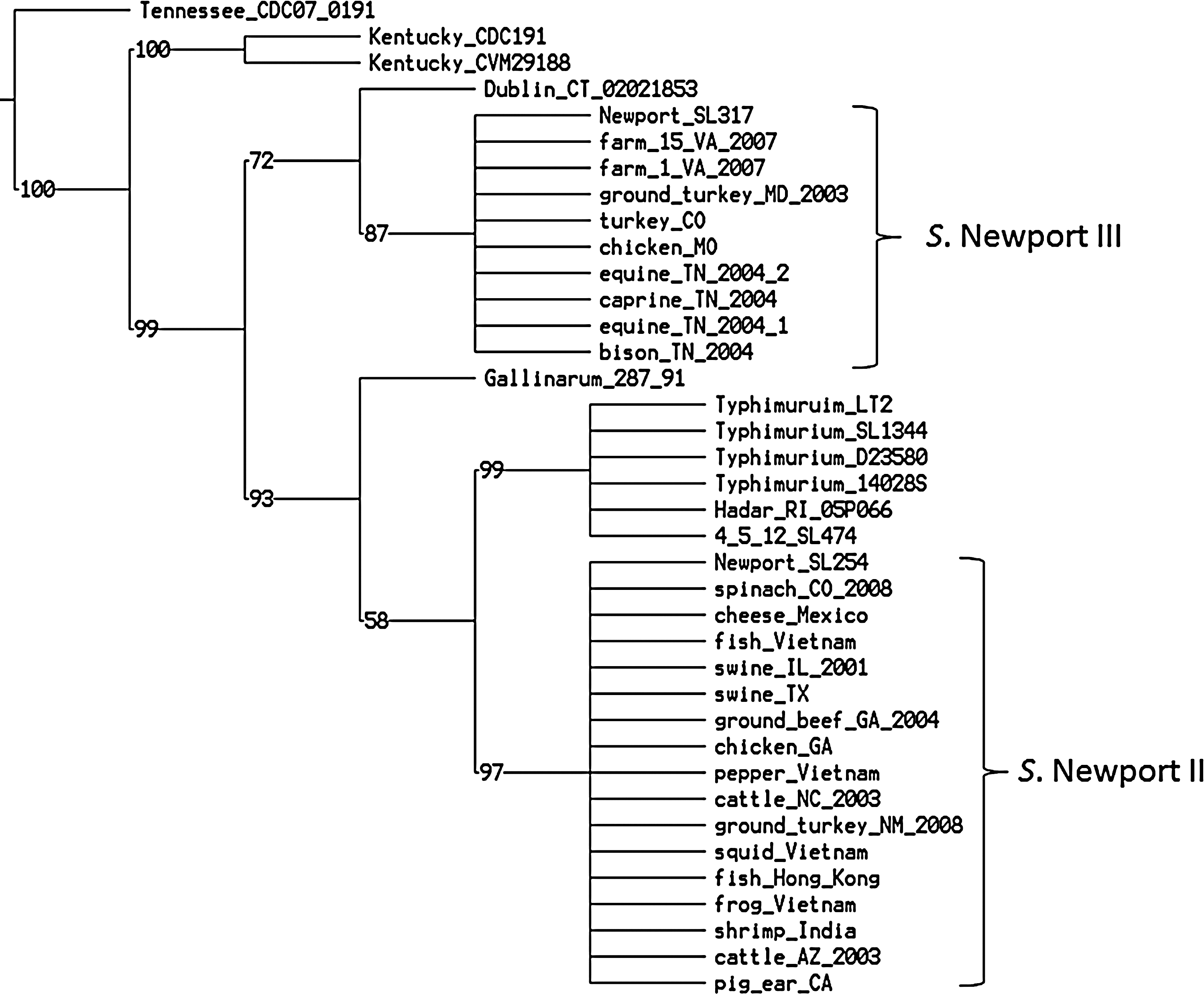
Parsimony phylogenetic tree of SPI-5 genes. There are 227 equally most parsimonious trees identified with a length of 187 single nucleotide polymorphisms, and consistency index of 0.797, and retention index (RI) of 0.942. Lineages II and III were separated by outgroup genomes. Lineage II displays close relationship with Salmonella Typhimurium group, Salmonella 4,[5],12:i:- SL474, Salmonella Hadar RI_05P066, and Salmonella Gallinarum 287/91; lineage III shows a close relationship with Salmonella Dublin CT_02021853.
Distances were calculated using the concatenated alignment of single nucleotide polytmorphisms in SPI-5 and saf fimbrial operon that estimate the diversity between two major lineages and outgroup genomes observed. Standard deviation is listed in parentheses.
Shrimp_India and Pig_ear_CA are Salmonella Newport strains and not included in the group Newport IIA&B and Newport IIC.
A total of 39 SNPs in SPI-5 defined lineages II and III, meaning that all strains in each lineage shared the same nucleotide sequence (4 SNPs in pipA, 9 in pipB, 7 in pipC, 5 in sopB, and 14 in pipD) (Table 3). Among the lineage-defining SNPs, 18 SNPs led to nonsynonymous substitutions, including 4, 7, 2, 2, and 3 nonsynonymous substitutions in pipA, pipB, pipC, sopB, and pipD, respectively.
A total of 39 SNPs in five genes in SNP-5 were identified. They defined Salmonella Newport lineages II and III, and could be used as potential biomarkers to differentiate strains during outbreak trace-back investigations. There are a total 18 SNPs causing nonsynonymous substitutions.
Genetic diversity of Salmonella pathogenicity island 6
An intact SPI-6 (T6SS part and saf operon) was present in all Salmonella Newport genomes, except the Asian strains in subgroup IIA including shrimp_India, squid_Vietnam, and pepper_Vietnam (Fig. 1). These three Asian strains contained one common gene cluster, named SPI-6 genomic island 1 (SPI6-GI1), and the saf genes. Thus, the “T6SS part” and the saf genes were analyzed separately. The complete genome Salmonella Virchow SL491 had gene contents identical to those of these Asian strains. Thus, Salmonella Virchow SL491 was used as an example to show the genetic characterization of SPI6-GI1 (Supplementary Table S4). According to the annotation, SPI6-GI1 did not carry any gene known to be related to virulence or antimicrobial resistance.
The phylogenetic tree based on T6SS was constructed using 937 SNPs (Fig. 3). TNT program identified 208 equally parsimonious trees with the same taxa at the subgroup level. The tree reflected the geographic origin of the isolates at the lineage level, meaning that the Asian strains in subgroup IIB were clustered separately from all American strains. Among the American strains, lineage III and subgroup IIC were separated. There were 672 SNP differences between IIB and IIC, but only 222 between IIB and Salmonella Hadar RI_05P066.
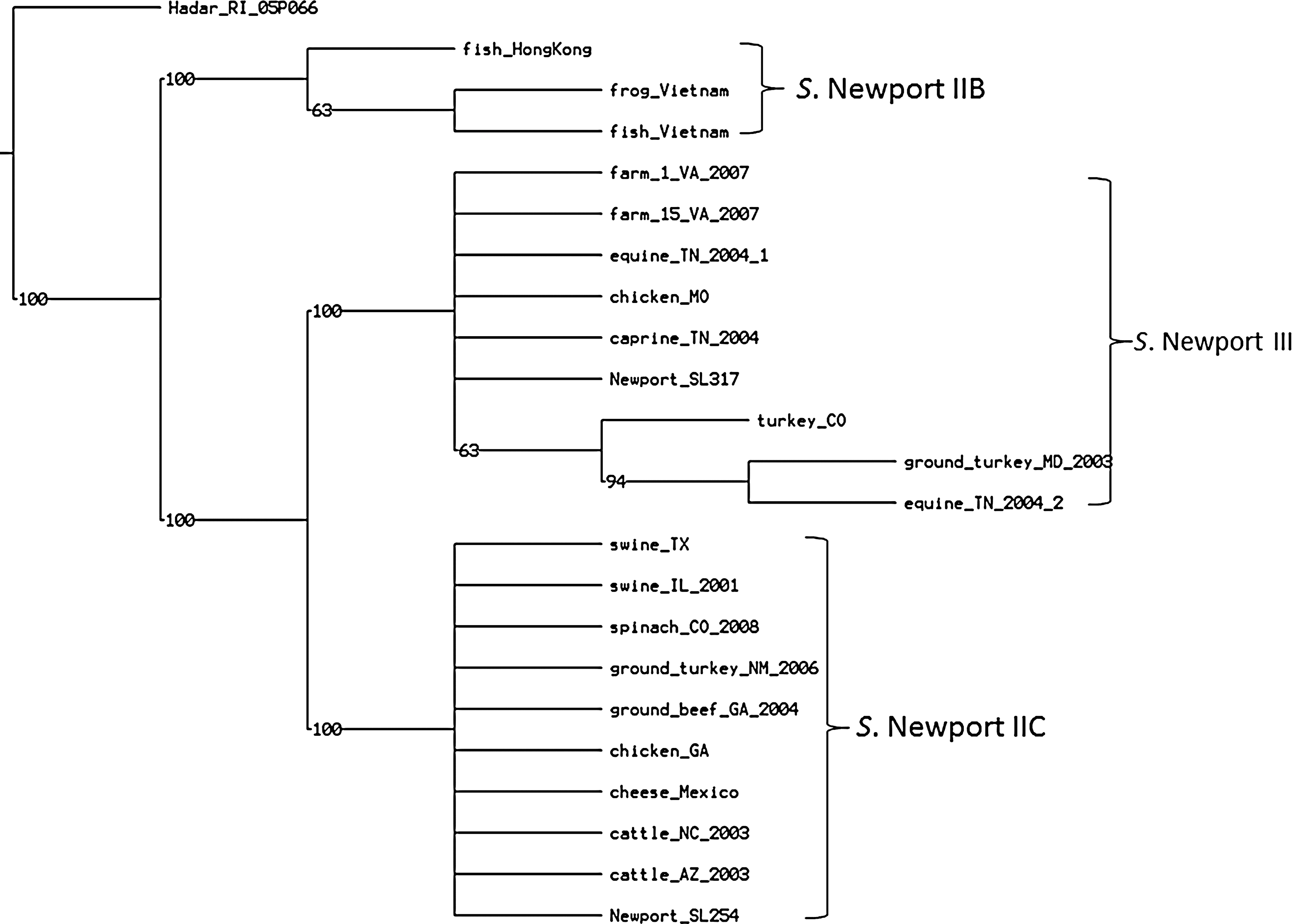
Parsimony phylogenetic tree of SPI-6 genes. There were 208 equally most parsimonious trees determined with a length of 1029 single nucleotide polymorphisms, consistency index of 0.914, and retention index (RI) of 0.984. SPI-6 clustering reflects geographic origins. The Asian strains were clustered separately from the American strains.
All 28 Salmonella Newport strains contained the safABCD operon. Similar to the T6SS tree, the six Asian strains were grouped together and clustered separately from the American strains (Fig. 4). Subgroup IIB strains clustered together. Strain shrimp_India displayed a distant relationship with the other five Asian strains. Salmonella Tennessee contained 71 SNP differences with IIA&B and only 21 SNP differences with shrimp_India (Fig. 4, Table 2). In the American group, lineage III and subgroup IIC were separated by Salmonella Gallinarum 287/91. Strain pig_ear_CA in IIA seemed to be an exception, showing a close relationship with lineage III (Table 2). Additionally, a gene cluster consisting of the tcfABCD fimbrial operon, tinR, and tioA, was only present in strains squid_Vietnam and pepper_Vietnam in subgroup IIA (Supplementary Fig. S2).
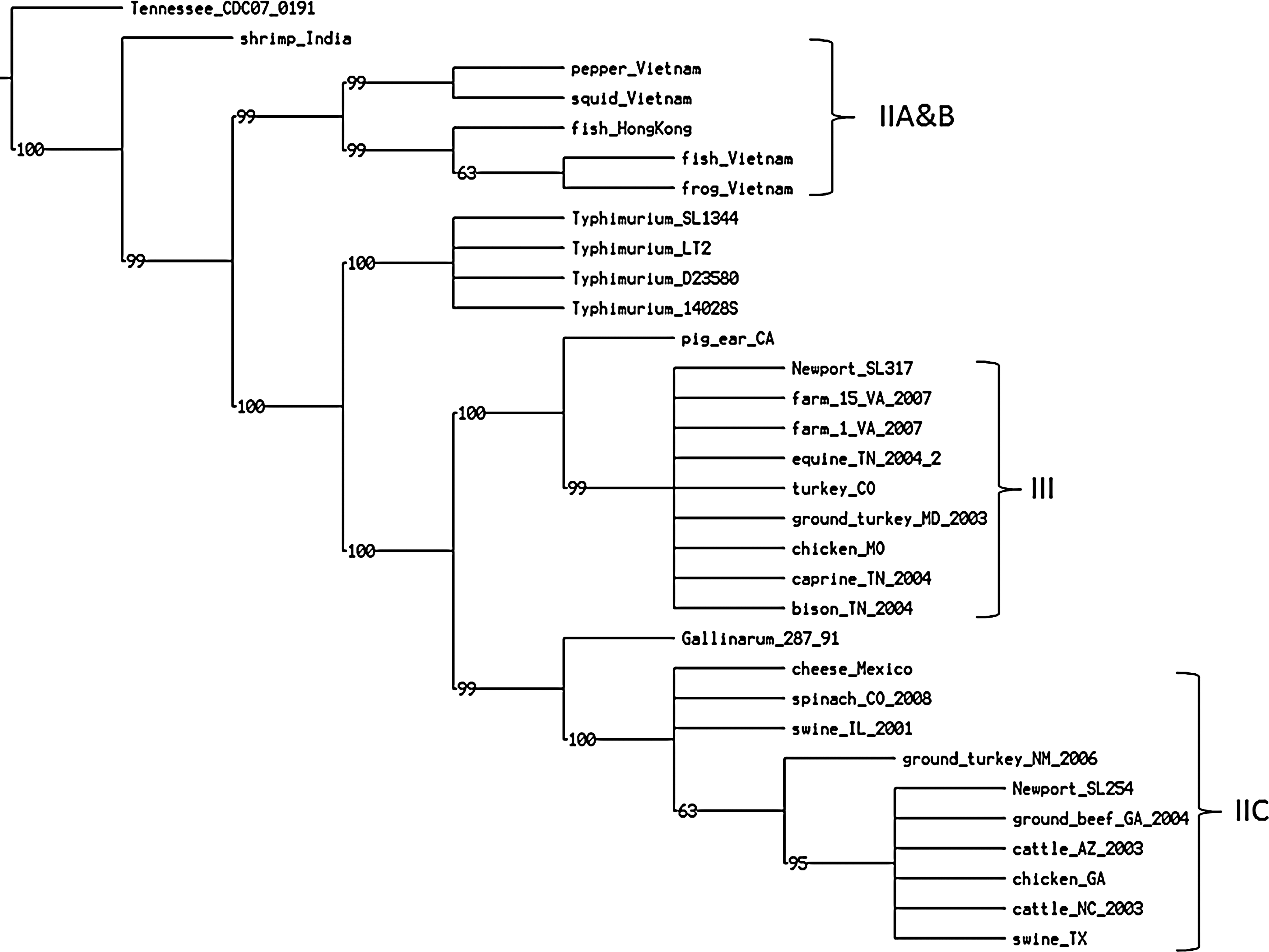
Parsimony phylogenetic tree of saf gene cluster. There are 210 equally most parsimonious trees determined with a length of 493 SNP, consistency index of 0.840, and retention index of 0.970. The saf genes clustering reflect geographic origins. All Asian strains were clustered separately from the American strains.
Discussion
SPIs play significant roles in causing human illness (Sabbagh et al., 2010). Due to the similarities in nucleotide sequence between bacteriophages and pathogenicity islands (PAIs), PAIs likely originated from phage via horizontal gene transfer (HGT). Examples include SPIs (Sabbagh et al., 2010), Vibrio pathogenicity island, and Staphylococcus aureus pathogenicity island 1 (SaPI1) (Boyd et al., 2001). Knodler et al. (2002) reported that SPI-5 genes might have been acquired through HGT from lambdoid phages, including Gifsy-1 and Gifsy-2. Therefore, bacteriophages may play vital roles in virulence activities of Salmonella and facilitate survival of the bacteria in different environments. For example, bacteriophages have been important for the genomic evolution of Salmonella Montevideo and Salmonella Enteritidis (Allard et al., 2012, 2013).
SPI5-GIs containing bacteriophage genes also may play significant roles in virulence. We hypothesized that SPI5-GI1 was originally acquired by the most recent common ancestor of node M via HGT and transmitted it vertically to the offspring strains (Fig. 1). SPI5-GI1 may have become functionally compatible with the genomes in node M, which includes all MDR strains (Cao et al., 2013). The presence of SPI5-GI2 indicates that the location between tRNA-ser and pipA may be a hot spot for independent acquisitions of foreign genetic elements. Functional studies of SPI-5 with and without these GIs might be important to examine the possible role of both SPI5-GIs. Both SPI5-GIs contained genes encoding a methylase, which could potentially regulate chromosome replication, cell cycle events, pathogenicity, and gene expression (Fang et al., 2012; Davis et al., 2013).
The SPI-5 genes could be considered targets for resequencing and biomarkers to rapidly differentiate lineages II and III. We performed positive selection tests for pipA and pipB using codon-based Z tests in MEGA6, indicating that these two genes were under positive selection. Positive selection played critical roles in the evolution of bacterial pathogens in that it accounts for 1.2% of the Salmonella core genome including virulence genes (Soyer et al., 2009). Soyer et al. reported that three genes showed evidence of positive selection in SPI-1 through SPI-6, including pipB (SPI-5) and safC (SPI-6) (Soyer et al., 2009). Since Salmonella Newport and pipA were not included in Soyer's study, pipA may show serotype-specific positive selection in Salmonella Newport.
Nonsynonymous substitutions in SPI-5 may have influenced the pathogenicity of the corresponding isolates. Two nonsynonymous substitutions were identified in domain CHASE3 in pipA, which is associated with signal transduction pathways in bacteria (Zhulin et al., 2003). pipB encodes a translocated effector of T3SS in SPI-2 (Knodler et al., 2002; Hensel, 2004). Moreover, a pipB null mutant caused reduced virulence in bovine hosts (Wood et al., 1998) and facilitates colonization of the cecum in chickens (Soyer et al., 2009). Nonsynonymous mutations were determined in the Chaperone_III domain in pipC, which is involved in T3SS and in delivering virulence effector proteins from Salmonella to host cells (Luo et al., 2001). The genes under positive selection could be possible targets for mutational studies (Soyer et al., 2009).
The three Asian strains in IIA may have different virulence attributes because they do not contain T6SS, which is a major component in SPI-6 (Jani and Cotter, 2010). Salmonella Gallinarum 287/91, Salmonella Virchow SL491, and Salmonella Paratyphi B SPB7 did not contain SPI-6 either (Blondel et al., 2009). Thus, the gain or loss of SPI-6 has occurred independently in different serotypes. We could not determine whether SPI6-GI1 was introduced independently or if it replaced T6SS. Since SPI-6 was located next to tRNA-asp and contained Rhs family protein genes, both of which are associated with rearrangement or acquisition of new genetic elements (Hill, 1999; Pukatzki et al., 2009), this location is likely to be a hot spot for recombination events. In the phylogenetic trees of SPI-6 and saf, the American strains in both lineages were clustered separately from the Asian strains, indicating that geographic location played an important role in the evolution and diversity of SPI-6. Based on the distribution of T6SS, saf, and tcf genes, the acquisitions of these clusters were independent events.
The findings in the current study distinguish Salmonella Newport lineages as well as MLST analyses (Cao et al., 2013). Moreover, no Salmonella Newport lineage I strain has been sequenced to date. The lineage I strains may possess the lineage-specific SNPs in SPI-5 and SPI-6 because lineage I displayed a distant relationship with lineages II and III (Sangal et al., 2010).
In addition, the tcf fimbrial operon, tinR, and tioA were only identified downstream of sinR in IIA, pepper_Vietnam, and squid_Vietnam (Supplementary Fig. S2). These genes were found downstream of SPI-6 in Salmonella Typhi, but not in Salmonella Typhimurium (Sabbagh et al., 2010). Porwollik (Porwollik, 2011) reported that Salmonella with a broad host range always possess higher numbers of fimbrial operons than those with host restriction. Diversification of the fimbrial operon in Salmonella may contribute to virulence activities (Yue et al., 2012; Allard et al., 2013). Moreover, the typhoid-associated gene tcfA has been more common in nontyphoidal Salmonella than known, and it is expressed during Salmonella invasion activities (Suez et al., 2013).
Conclusions
SPI-5 and SPI-6 possess extensive differences in Salmonella Newport lineages II and III. SPI-5 contained both insertions and substitutions, including SPI5-GI1 and SPI5-GI2. SPI6-GI1 was present in the Asian strains of IIA. These genomic islands may contribute to virulence in their hosts. The SNPs in SPI-5 and SPI-6 could be used as biomarkers for rapid detection and epidemiological investigations to differentiate Salmonella Newport lineages II and III. The tcf genes may relate to host range and virulence activity in pepper_Vietnam and squid_Vietnam.
Footnotes
Acknowledgments
This work was supported in part by the Joint Institute for Food Safety and Applied Nutrition, University of Maryland.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.